

Dopo aver introdotto brevemente i primi concetti sulle anemie in ICU ed aver parlato delle due forme di anemie più frequentemente riscontrate in clinica (microchimiche e macrobiotiche), in questo capitolo ci concentreremo sulle forme di anemie più specifiche. Le anemie emolitiche sono quel gruppo di anemie che risultano da un incremento del numero di eritrociti distrutti; generalmente i globuli rossi hanno un’emivita di 120 giorni, ma in questo caso con l’aumento della distruzione eritrocitaria la durata media si accorcia in maniera sensibile. Un midollo osseo normale è in grado di aumentare la produzione cellulare di 6-8 volte rispetto alla norma, per cui la distruzione emolitica deve essere veramente importante, con un’emivita media inferiore a 30 giorni per diventare clinicamente evidente.
Le anemie emolitiche si classificano a loro volta in ereditarie o acquisite. Le forme ereditarie sono legate ad un difetto eritrocitario intrinseco ed ereditario, che si può classificare in base alla sede d’interesse; può essere coinvolta l’emoglobina per cui si parla di emoglobinopatie (come la talassemia ed i difetti globinici), può essere coinvolto il metabolismo cellulare se si hanno alterazioni nelle funzioni enzimatiche dei globuli rossi (come il deficit di G6PDH e/o nella piruvato chinasi) oppure può essere coinvolta la membrana quando si hanno alterazioni nella struttura di membrana (in tal caso si parla di Sferocitosi, Ellissocitosi, ecc…). Le forme acquisite sono forme legate ad cambiamenti extracorpuscolari e/o ambientali; si classificano in base alla causa del danno; la causa può essere immunologica ed allora si parla di anemie immunoemolitiche da auto-anticorpi (auto-immune) o iso-anticorpi (allo-immune), la causa può essere la frammentazione ed in tal caso sono anemie dovute a graft arteriosi, valvolari, microtrombotici, ecc…, che provocano frammentazione eritrocitaria oppure possono essere con emoglobinuria, solitamente da infezione, epatopatie, nefropatie, ecc…; fra queste l’emoglobinuria parossistica notturna è l’unica eccezione di anemia dove si hanno delle forme acquisite ma su un deficit intrinseco al globulo rosso già pre-esistente.
DIAGNOSI:
La clinica delle anemie emolitiche è molto simile alle forme di anemia classica (si veda il capitolo precedente, Capitolo 2.10b.1-1) per cui esiste una sintomatologia aspecifica tipica dell’anemia cui si aggiunge una sintomatologia specifica delle forme emolitiche, con lieve ittero, lieve splenomegalia, rischio di colelitiasi (con calcoli pigmentati per accumulo di bilirubina), rischio di ulcere alle caviglie ed aumentato rischio di episodi di anemia aplastica (per co-infezione di Parvovirus B19). Al laboratorio si ha un aumento degli indici di emolisi con aumento notevole della bilirubina indiretta, dell’urobilinogeno urinario e dello stercobilinogeno fecale, con associata riduzione importante della aptoglobina (che generalmente arriva a 0). A questo si associano degli indici di danno cellulare con alterazioni morfologiche dei globuli rossi, riscontro di fragilità osmotica, ridotta emivita (se si misura l’emivita eritrocitaria con il Cromo radioattivo) e degli indici di maggiore eritropoiesi con reticolocitosi ed iperplasia eritroide midollare. Le alterazioni cellulari che si riscontrano in questi casi sono estremamente varie (si può avere sferocitosi, microsferocitosi, poichilocitosi, policromia, punteggiatura basofila, ecc…); il normale rapporto mieloide/eritroide solitamente nel midollo va da 2:1 a 12:1, mentre in caso di iperplasia eritroide midollare (come reazione all’aumentato catabolismo si ha un’inversione del rapporto, che va da 1:1 a 1:12.

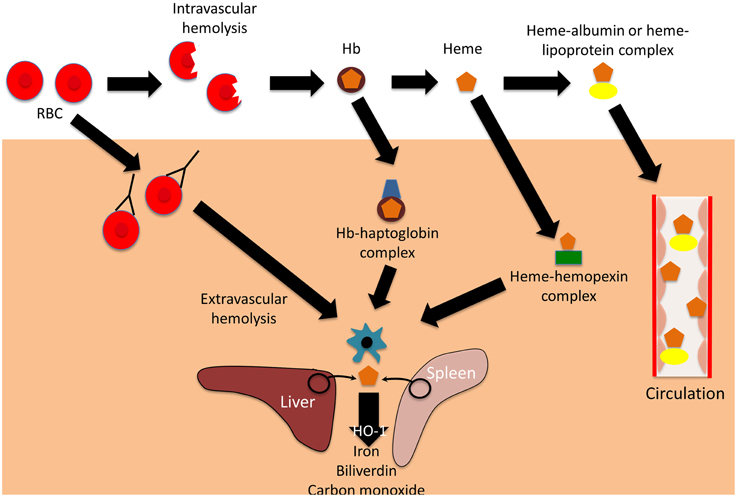
Per quello che riguarda poi la sede dell’emolisi (altro parametro importante per la diagnosi ed il monitoraggio clinico), la sede può essere extravascolare quando si ha un incremento della normale emocateresi splenica, con il riutilizzo efficace del materiale catabolizzato oppure intravascolare quando si ha una rottura diretta delle emazie nei vasi, con un rapido consumo di aptoglobina (con cui forma un legame irreversibile con un rapporto stechiometrico di 1:1), conseguente filtrazione glomerulare dell’emoglobina in eccesso che fino ad un determinato livello viene recuperata dal tubulo contorto prossimale renale, per poi essere escreta con l’urina (ed in questi casi si parla di emoglobinuria).
1) TALASSEMIE:
Le talassemie sono delle patologie ereditarie provocate da errori nella sintesi delle catene emoglobiniche; ne esistono diversi sottotipi, di cui i più importanti sono l’α/β-talassemia. Dal punto di vista eziopatogenetico il deficit nella sintesi delle catene globiniche può colpire ogni step nella sintesi proteica, ma fra tutti i quadri più frequenti sono per l’α-chain la delezione genomica, mentre per la β-chain sono la delezione genomica, le mutazioni non-senso o ancora altri meccanismi. La trasmissione ereditaria di tali geni segue le leggi di Mendel per entrambe le catene (sapendo che per la catena-α i due geni sono trasmessi come aplotipo). Nell’α-talassemia la delezione può colpire un solo gene (definito come α- o α+) o entrambi i geni (-- o α0) di un cromosoma (bisogna ricordarsi che ogni cromosoma presenta due geni α); si hanno pertanto diverse combinazioni possibili:
- αα/α-: definito come portatore silente (α/α+);
- α-/α-: definito come trait talassemico (α+/α+);
- αα/--: definito come trait talassemico (α/α0);
- --/α-: definito come malattia da emoglobinaH (α0/α+);
- --/--: idrope fetale, emoglobina di Barts (γ4);
Nelle β-talassemie la delezione (o altri meccanismi eziologici) possono colpire il gene β che è presente in singola copia per ogni cromosoma; si hanno forme β+ (con conseguente riduzione della sintesi di catene-β) e forme β0 (senza sintesi di catene-β), ottenendo quindi diverse combinazioni:
- β/β1: talassemia minor;
- β/β0: talassemia minor;
- β+/β+: talassemia intermedia/major;
- β0/β0: talassemia major;
FISIOPATOLOGIA:
La talassemia comporta uno squilibrio nel rapporto normale che esiste fra catene α e non-α; solitamente tale rapporto è di 1:1, nelle β-talassemie eterozigoti si arriva ad avere un rapporto α:β di 2:1, mentre nelle β-talassemie omozigoti si arriva anche a 5-25:1. La catena in eccesso si accumula a livello intracellulare, eccedendo le normali capacità della cellula di degradazione proteica, generando danni intracellulari. In microscopia elettronica è visibile difatti un accumulo nucleare e citosolico di catene globiniche, che aumenta sempre di può con la maturazione eritroblastica (si parla di Corpi di Heintz).
Gli effetti cellulari sono un’eccessiva liberazione di radicali liberi dell’ossigeno, con perossidazione della membrana, formazione di metemoglobina e danno delle differenti vie metaboliche cellulari, cui segue la morte cellulare pereritropoiesi inefficace, con blocco cellulare in fase G1 e morte cellulare midollare. Inoltre si associa un’alterazione nella normale fosforilazione delle proteine di membrana, con conseguente poichilocitosi. Gli effetti sistemici sono una riattivazione della produzione di γ/δ-chain, con formazione di emoglobina-A2 ed emoglobina-F, che risultano efficaci nelle forme lievi di talassemia. Nel tempo si ha una chiara anemia per eritropoiesi inefficace ed emocateresi splenica incrementata (come conseguenza della microcitosi, dell’ipocromia e della anisopoichilocitosi cellulare); per aumentare l’eritropoiesi il midollo osseo si espande nelle ossa, si riattiva nella milza, nel fegato ed in altre aree (soprattutto nella regione paravertebrale). La milza va incontro a splenomegalia per aumento dell’emocateresi dei globuli rossi lesionati; il conseguente sequestro locale delle red cells comporta maggiore produzione di emazie (tutte alterate), con formazione di un circolo vizioso. Infine si ha un accumulo marziale nell’organismo dovuto ad un processo emolitico con siderosi che si manifesta soprattutto al miocardio, al fegato ed al pancreas.

CARATTERISTICHE DELLE β-TALASSEMIE:
La β-talassemia major detta anche morbo di Cooley è la forma più grave di talassemia, con sintomatologia evidente dal IV-VI mese di vita (nel momento in cui compare lo shift di catena fra quelle γ e quelle β). Clinicamente si ha ipoevolutismo somatico (basso peso ed altezza), facies mongoloide, cute pallida, ittero e danno d’organo (con cardiomegalia, splenomegalia massiva ed epatomegalia). Il bambino muore se non viene adeguatamente trattato (mediante trasfusione e rimozione del ferro) entro pochi mesi. Nella β-talassemia intermedia i pazienti presentano un quadro clinico-ematologico a metà fra la forma major e minor, solitamente generata da forme β+/β+, raramente da forme β+/β0 con maggiori capacità di degradazione dell’α-chain. In questi bambini si ha un normale sviluppo psicofisico, ma pallore cutaneo (di intensità variabile), epatosplenomegalia (con siderosi epatica), colelitiasi ed ulcere trofiche malleolari (relativamente rare, ma sono state descritte). Nella β-talassemia minor detta anche malattia di Rietti-Greppi-Micheli il paziente è un soggetto cronicamente anemico fin dall’infanzia, con una malattia minimamente visibile (a volte viene riscontrata occasionalmente). In questi pazienti si hanno segni/sintomi di anemia, subittero e splenomegalia. Infine il trait talassemico detto anche β-talassemia minima si caratterizza per un’eterozigosi β+/β senza manifestazioni cliniche di malattia. La diagnosi viene posta al laboratorio con riscontro di poliglobulia, microcitosi ed ipocromia; si ha un incremento della emoglobina-A2.
Al laboratorio le β-talassemie mostrano all’emocromo anemia (per riduzione dell’emoglobina e dei globuli rossi), microcitosi (con MCV inferiore ai 80 fl), ipocromia (con MCH e MCHC ridotti), estrema anisopoichilocitosi (nella forma major), incremento degli eritroblasti periferici, leptociti (nella forma major) descritti come globuli rossi molto appiattiti che sono più resistenti alla lisi ipotonica in NaCl. A questo si associa un aumento della bilirubina diretta, riduzione dell’aptoglobina ed incremento della sideremia. Alla radiografia ossea si dimostra un assottigliamento della corticale in maniera generalizzata, con alterazione delle trabecole ossee craniche (con un aspetto che viene descritto a spazzola), e delle vertebre (con un reticolo descritto a ragnatela).
La prognosi delle β-talassemie per la forma major la malattia é severa, con un’anemia cronica e spiccata e la storia naturale vede una morte pre-adolescenza: il paziente sopravvive mediante emotrasfusioni (che comunque generano emocromatosi secondaria con danni d’organo importanti); per la forma Intermedia il tasso di sopravvivenza è variabile e la sopravvivenza può essere lunga anche in assenza di trasfusioni.
CARATTERISTICHE DELLE α-TALASSEMIE:
In quelle forme dove si ha l’emoglobina Barts composta da quattro catene γ, che sono catene ad elevata affinità per l’ossigeno; queste risultano inefficaci e poco stabili, per cui si ha una morte intrauterina del feto con idrope fetale. In quelle forme dove si ha l’emoglobina-H (tetramero di catene β) la forma clinica molto simile alla β-talassemia major, anche se frequentemente si hanno manifestazioni minori (con una splenomegalia ridotta rispetto alla forma major). Il trait talassemico ed il portatore sano sono invece asintomatici dato che raramente danno manifestazioni cliniche di anemia. Alle analisi di laboratorio le α-talassemie mostrano un emocromo con anemia ipocromica e microcitica (di diversa severità), anisopoichilocitosi e precipitazione dell’emoglobina-H (di colore blu brillante cresile).
DIAGNOSI TALASSEMIE:
Il sospetto diagnostico si pone con l’anamnesi e l’esame obiettivo. (che dimostrano la presenza di anemia), mentre la certezza diagnostica si ottiene mediante le indagini di laboratorio con l’emocromo, lo striscio di sangue periferico e l’elettroforesi dell’emoglobina. Nei pazienti omozigoti la diagnosi è semplice, mentre nei pazienti eterozigoti si malattia si pone in diagnostica differenziale con l’anemia sideropenica (microcitica ed ipocromica), che si differenzia in ultima analisi per una bassa sideremia ed un basso numero di reticolociti.
TERAPIA:
Le emotrasfusioni sono necessarie per le forme molto gravi di malattia, anche se possono essere necessarie in qualche forma intermedia di malattia; lo scopo è quello di mantenere i valori di emoglobina attorno a 10-12 g/dl, così da bloccare l’iperplasia midollare eritroide e l’eritropoiesi inefficace. In questi pazienti é importante co-somministrare dei chelanti del ferro (come la Desferoxiamina mesilato, 40 mg/Kg die in infusione SC) per ridurre l’emocromatosi secondaria; generalmente la terapia si inizia dopo 50 UI di emazie trasfuse. Gli effetti collaterali possono essere immediati come irritazione cutanea locale, prurito e/o ipotensione arteriosa, piuttosto che tardivi con neuropatia ottica, deficit acustico neurosensoriale o elevato rischio infettivo.
La splenectomia si esegue nei pazienti che subiscono numerose trasfusioni e/o presentano una grave splenomegalia (di grado IV); è meglio eseguirla dopo i 5 anni d’età, perché si evita il rischio di setticemie fatali (soprattutto da S. pneumoniae, H. influenzae e N. meningitidis). L’acido folico si somministra per sopperire alla carenza di folati, condizione tipica delle anemie emolitiche per espansione della massa eritropoietica.
2) EMOGLOBINOPATIE:
Le emoglobinopatie sono delle condizioni ereditarie caratterizzate dalla sintesi di emoglobina strutturalmente anomala; ne esistono circa 400 varianti polimorfiche, di cui solo 1/3 risultano patologiche; le mutazioni possono essere puntiformi (condizione più frequente), con allungamento di catena e/o per fusione di geni differenti. Esistono veramente numerose tipologie di emoglobinopatie, alcune senza alterazioni funzionali (come l’emoglobina-G Philadelphia), alcune da aggregazione (come l’emoglobina-S, l’emoglobina-C, l’emoglobina-D o l’emoglobina-E), altre instabili (come l’emoglobina Koln), la Metaemoglobina (detta anche emoglobina-M) o altre ancora con differente affinità per l’ossigeno (in eccesso/difetto). La trasmissione genica segue sempre le leggi di Mendel, con manifestazioni codominanti, dominanti, recessive, ecc…
SINDROMI FALCEMICHE:
Le sindromi falcemiche sono condizioni clinico-patologiche estremamente eterogenee, legate alla presenza di un’emoglobina anomale, che si caratterizzano per mutazioni puntiformi intracatena: l’emoglobina-S (sickle cell disease) vede una mutazione sulla catena β in posizione 6 fra Glutammato e Valina (β6 Glu-Val), l’emoglobina-C anch’essa vede una mutazione sulla catena β in posizione 6 fra glutammato e Lisina (β6 Glu-Lys), l’emoglobina- D in posizione 121 fra Glutammato e Glutammina (β121 Glu-Glutamina) ed infine l’emoglobina-E in posizione 26 fra Glutammato e Lisina (β26 Glu-Lys). L’emoglobina-S è un polimorfismo bilanciato che risulta vantaggioso in alcune aree, soprattutto dove la malaria è endemica ed é endemica in alcune aree, soprattutto in Africa, in India e nel bacino del Mediterraneo arrivando in alcune aree ad essere presente fino al 10-40% della popolazione. L’emoglobina-C invece è diffusa soprattutto in Africa (40%), mentre l’emoglobina-D é più rappresentata in India (3%) e l’emoglobina-E nel Sud-Est asiatico (30%).
Per quello che riguarda la patogenesi, nel caso si abbia una tensione di ossigeno bassa (inferiore a 50-60 mmHg) si ha un’adesione dell’emoglobina-S, con formazione di fibre tubulari (chiamate fibre tattoidi) a 6-8 filamenti disposti in spirale con cui si crea una struttura rigida, che deforma la cellula, portando alla formazione di Drepanociti. Ciò che può indurre i drepanociti sono pertanto una bassa tensione di ossigeno (inferiore a 50-60 mmHg), la disidratazione, le alte temperature (febbre) ed un tasso di emoglobina-S superiore al 60%. Le alterazioni morfologiche sono reversibili nei drepanociti giovani, mentre rimangono irreversibili nei drepanociti più anziani; sono cellule che hanno purtroppo una breve emivita (per una facile emolisi intra/extravascolare) ed una maggiore tendenza all’aggregazione (sull’endotelio, sui globuli bianchi, ecc…). A livello fisiopatologico i drepanociti vengono quindi intrappolati nel microcircolo (soprattutto nella milza), l’ipossia stimola la produzione di Epo, che porta iperviscosità, incrementando l’ostacolo al circolo sanguigno e generando un circolo vizioso. Le alterazioni fisiopatologiche più comuni che si vengono a riscontrare sono delle sindromi vasculolesive, delle sindromi dolorose (da ipossia), un’emolisi intravascolare ed un incremento dell’adesione intercellulare (nelle aree a basso flusso).
Clinicamente esistono numerosi quadri clinici, soprattutto evidenti in caso di omozigosi S/S ed eterozigosi fra le altre forme patologiche. La malattia rimane generalmente asintomatica e si caratterizza per degli episodi (più o meno frequenti) di crisi occlusive che possono verificarsi a livello cardiaco (con ischemia, infarto miocardico, ecc…), a livello polmonare (dove sono estremamente frequenti tali lesioni, con progressiva alterazioni del rapporto V/Q, si veda il capitolo dedicato, Capitolo 3.1.4), a livello renale (con ematuria non dolorosa secondario all’ischemia glomerulare) a livello osseo con infarti ossei, osteomieliti che possono sfociare nell’Hand-Foot disease con microinfarti del carpo, mani edematose e lesioni ossee, negli occhi con degenerazion retinica, microaneurismi e nella cute con ulcere trofiche degli arti inferiori. Le altre crisi importanti sono crisi dolorose legate all’occlusione vascolare acuta da parte dei drepanociti bloccati nel microcircolo; le cause sono le stesse che provocano drepanocitosi e la sintomatologia legata al sequestro cellulare con splenomegalia, soprattutto evidente durante l’infanzia; a volte si ha anche sequestro epatico/polmonare. Infine sono purtroppo più frequenti rispetto alla popolazione sana delle crisi aplastiche con anemia acuta da switch-off dell’eritropoiesi, con una riduzione dei reticolociti; tipicamente un fattore scatenante é la co-infezione da parte del Parvovirus B19.

Alle analisi di laboratorio si hanno le alterazioni tipiche delle anemie emolitiche (vedi sopra); per la diagnosi non si utilizza l’elettroforesi emoglobina perché le emoglobine anomali non dimostrano una differente migrazione rispetto all’emoglobina classica, ma il test di falcizzazione: mediante l’uso di una sostanza riducente miscelata al campione si valuta se si formano i drepanociti, che risulta essere un test diagnostico.
Per quello che concerne la prognosi con una buona nutrizione ed un controllo delle infezioni il paziente solitamente rimane senza lesioni gravi; a volte può mostrare un quadro lieve di pubertà ritardata. I pazienti con omozigosi S/S di contro raramente superano i 50-60 anni d’età per la comparsa di complicazioni legate all’anemia, all’accumulo di ferro o alle trasfusioni. Purtroppo non esiste una terapia specifica in grado di migliorare le manifestazioni cliniche; le crisi dolorose possono essere controllate con idratazione IV ed analgesici, oltre ad arrestare la causa evidente (se si ritrova) della generazione drepanociti.
COMPLICAZIONI LEGATE AL TRAIT FALCEMICO:
Il trait falcemico è una condizione caratterizzata dalla presenza di un gene emoglobinico normale associato ad un gene di emoglobina β1-mutato; essendoci 4 tipologie differenti di gene β1, la tipologia di gene ereditato determina la severità della condizione clinica. La malattia affligge circa 300 milioni di persone nel mondo, con una maggiore concentrazione in Africa e nell’area Mediterranea, dove la presenza della malaria (a cui il trait falcemico è più resistente al P. falciparum, ma non agli altri plasmodi) ha determinato una selezione positiva di tali soggetti.
Tradizionalmente si pensa al trait falcemico come una condizione benigna, dato che conferisce una resistenza all’infezione da P. falciparum, senza alcuna manifestazione clinica di malattia. I pazienti con trait falcemico hanno valori di emoglobina e di ematocrito sovrapponibili ai valori della popolazione sana, ma la condizione non è totalmente benigna, dato che stanno progressivamente aumentando i riscontri in letteratura sulle patologie associate al trait falcemico. La condizione fisiopatologica è secondaria alla polimerizzazione della emoglobina-S, che comporta una successiva deformazione dei globuli rossi, condizione favorita dall’ipossia, dall’acidosi, dall’incremento della viscosità, dalla disidratazione e dall’ipertermia.
Sono presenti delle associazioni certe con alcune malattie. Il carcinoma renale midollare é una tipologia di tumore renale raro e particolarmente aggressivo che generalmente si manifesta nella popolazione giovanile con trait falcemico, generalmente con una preponderanza maschile (3:1) prima dei 24 anni e dopo questa età con uguale frequenza nei due sessi. Il tumore origina dall’epitelio dei dotti collettori distali e cresce in maniera infiltrativa, invandendo i sinusoidi renali; dal punto di vista citologico si tratta di un gruppo di cellule coese con citosol vacuolato con modificazioni geniche abbastanza specifiche. Dal punto di vista clinico la malattia diviene sintomatica per la presenza di dolore e peso a livello del fianco interessato dal tumore, generalmente associato ad ematuria; la diagnosi si pone mediante TC/RMN-addome ed analisi istologica. La prognosi è pessima, con una mediana di sopravvivenza di 15 mesi, e la terapia è tradizionalmente la nefrectomia radicale con chemioterapia adiuvante e/o radioterapia palliativa.
La necrosi papillare renale é una condizione clinico patologica che rappresenta il 4% dei ricoveri nella popolazione con trait falcemico, necrosi secondaria alla formazione di microinfarti midollari renali dove si ha iperviscosità, acidosi, ipossia, ecc… che promuovono la polimerizzazione della emoglobina-S. La manifestazione clinica della malattia è tipicamente l’ematuria, dove generalmente il rene sinistro è il più coinvolto per una maggiore pressione venosa secondaria alla compressione sulla Vena Renale da parte della pinza aorto-mesenterica-superiore. La diagnosi si pone mediante riscontro di frazioni di papille renali nelle urine (condizione patognomonica, ma rara), e più frequentemente mediante cistouroscopia, TC-addome, Ecografia addome. Il trattamento per le forme lievi-moderate è solitamente di tipo conservativo con riposo a letto, idratazione IV ed alcalinizzazione delle urine, mentre per le forme severe si esegue un tamponamento uroscopico diretto, con eventuale somministrazione di desmopressina acetato. Eventuali complicanze sono infezioni delle vie urinarie e/o la comparsa di nefrolitiasi.
L’ipostenuria si tratta dell’incapacità del rene di poter concentrare le urine ed è una condizione secondaria a ripetuti microinfarti midollari renali, che comporta una importante perdita di acqua libera che, se non compensata, può portare ad un incremento della Osmolarità plasmatica. Gli infarti splenici sono complicanze estremamente frequenti in questi pazienti, soprattutto in condizioni in cui si hanno basse tensioni locali di ossigeno; clinicamente possono essere asintomatiche, oppure manifestarsi con segni di irritazione peritoneale e/o irritazione pleurica (con versamento pleurico sinistro ed atelettasia reattiva); la splenectomia è indicata soprattutto in caso di rotture spleniche con sanguinamento peritoneale importante, ascessi splenici e/o splenomegalie massive. Infine le morti esercizio-indotte sono condizioni rare, anche se occorrono più frequentemente del solito nella popolazione affetta da trait falcemico (circa 28 volte rispetto alla popolazione sana) scatenato dall’esercizio fisico che comporta disidratazione, ipertermia, acidosi metabolica, ipossia ed ipostenuria che favorisce la polimerizzazione dei globuli rossi, con occlusioni vascolari multiple, rabdomiolisi, DIC e vasospasmi coronarici. La terapia in questi casi è soprattutto di tipo preventivo, con idratazione di acqua e sali minerali, cessazione dell’attività fisica alla comparsa di dolori muscolari, somministrazione di ossigeno ed utilizzo appropriato di defibrillatori impiantabili.
Fra le associazioni probabili al trait talassemico alcuni studi eseguiti sulla popolazione nera afro-americana con trait falcemico hanno riscontrato un incremento del rischio di trombosi venosa profonda (2 volte rispetto alla norma) e di embolia polmonare (4 volte rispetto alla norma) con riscontro di livelli sierici più elevati di D-dimeri, dei complessi trombina-antitrombina, dei frammenti di protrombina e monocitosi. Si riscontra inoltre un maggiore tasso di aborti (9,7% vs 3,5%) secondariamente ad uno stato ipercoagulativo, ad anomalie placentari e ad anomalie fetali. Ad oggi viene consigliata una terapia preventiva in caso di procedure chirurgiche ortopediche, ginecologiche ed urologiche con eparina non frazionata. L’ifema si tratta invece di una condizione in cui all’interno dell’umor acqueo si riscontrano eritrociti, generalmente secondario ad un eccessiva stagnazione di red cells ed ipossia, con conseguente stagnazione di red cells a livello delle trabecole di meshwork. La condizione porta ad emorragie intraoculari, incremento della pressione intraoculare e rischio di occlusione arteriosa retinica, con conseguente atrofia del nervo ottico. La terapia è quella di abbassare la pressione intraoculare mediante l’utilizzo di ß-agonisti, steroidi topici e lavaggi della camera anteriore (più raramente con camera iperbarica). Sono da evitare farmaci come Acetazolamide e/o diuretici osmotici.
Fra le associazioni possibili esistono altre condizioni clinico-patologiche che mostrano qualche associazione con il trait talassemico, fra cui le retinopatie (soprattutto in pazienti con diabete mellito), le sindromi coronariche acute e/o le batteriurie asintomatiche.
3) ALTERAZIONI DEL METABOLISMO DEI GLOBULI ROSSI:
Queste patologie sono anemie emolitiche ereditarie secondarie alla riduzione di attività di alcuni enzimi necessari al metabolismo glucidico eritrocitario; le condizioni epidemiologicamente più frequenti sono il deficit di G6PD (Glucosio-6-fosfato deidrogenasi) e di PK (protein-kinasi). Le alterazioni metaboliche presentano una prevalenza di di circa 100 milioni di pazienti, soprattutto nelle aree del Mediterraneo, Africa, Asia minore, ecc… (del tutto sovrapponibili alla talassemia, alla malaria, alla drepanocitosi, ecc…). Il gene responsabile di malattia è sul cromosoma X, per cui la malattia è sintomatica nei maschi, mentre nelle donne dipende dal grado di inattivazione.
Le mutazioni genetiche comportano una ridotta efficienza enzimatica per la presenza di isoenzimi con caratteristiche biologiche e cinetiche alterate; la forma B+ é l’enzima più comune, con un’attività al 100%, presente nella razza caucasica, la forma A+ é il secondo enzima frequente, con un 84% di attività (presente nella razza afroamericana), che presenta anomalie elettroforetiche rispetto alla forma wild type. La forma A- é un enzima presente nella razza africana, che mostra un’attività del 5-15% e la forma mediterranea si caratterizza per un deficit di sintesi enzimatica (con un’attività allo 0%).
FISIOPATOLOGIA:
In un paziente sano con un globulo rosso normale il funzionamento della G6PD decresce lentamente; a 120 giorni funziona poco, ma ancora abbastanza per mantenere nella cellula buone concentrazioni di NADPH e GSH. In un paziente con deficit enzimatico si rileva (seppure con diversa velocità) un deficit progressivo di NADPH e GSH, con conseguente incremento di radicali liberi dell’ossigeno e ferro allo stato ferrico: l’emoglobina precipita (formando i corpi di Heintz) e provoca emolisi extravascolare. L’emivita delle emazie si riduce a 20 giorni.

CLINICA:
La sintomatologia del paziente é riconducibile ad episodi di emolisi acuta scatenate da infezioni o dall’uso di farmaci che si rilevano generalmente dopo 1-3 giorni con febbre, nausea, vomito, dolori addomino-lombari ed ittero. In altre forme (quelle più severe) si può avere emolisi cronica con una clinica analoga alle altre anemie emolitiche croniche, caratterizzate da ittero, anemia e manifestazioni acute legate alla presenza di fattori scatenanti. In altri pazienti si può avere ittero neonatale che rappresenta la causa più frequente di ittero neonatale dopo la malattia emolitica del neonato (a patogenesi alloimmune). infine va menzionato e ricordato il favismo, condizione che si manifesta clinicamente dopo assunzione di Vicia Flavia; colpisce soprattutto alcuni portatori della variante Mediterranea. Dopo 2 giorni dall’assunzione di fave il paziente manifesta emolisi acuta, che può anche portare alla morte; non è chiaro perché solo alcuni pazienti manifestino tale clinica, probabilmente sono coinvolti altri geni attualmente non identificati.
La diagnosi avviene mediante la clinica che pone il sospetto diagnostico (con il riscontro di anemia acuta/cronica) così come le analisi di laboratorio con dimostrazione dell’emolisi extravascolare; l’analisi diagnostica é la spettrofotometria che valuta direttamente l’attività enzimatica della trasformazione di NADP in NADPH.
La terapia purtroppo é misera; esiste una terapia patogenetica che cerca di evitare l’insorgenza di infezioni, l’uso di farmaci scatenanti e l’assunzione di fave. Per il resto le altre terapie son oda riservare ai casi più severi: la splenectomia non è sempre efficace e funziona per i quadri importanti di anemia emolitica extravascolare cronica e le trasfusioni vanno eseguite solamente nelle crisi acute.
4) ALTERAZIONI CITOSCHELETRICHE DEI GLOBULI ROSSI:
Le alterazioni citoscheletriche dei globuli rossi sono alterazioni genetiche primitive delle proteine costituenti lo scheletro di membrana, che comporta alterazioni funzionali cellulari che portano infine ad una riduzione dell’emivita. La patologia più frequente a livello epidemiologico è la Sferocitosi Ereditaria. Studi effettuati durante la lisi ipotonica dei globuli rossi (con rimozione dell’emoglobina) permette di ottenere delle strutture chiamate ghost perché i globuli rossi vuoti appaiono simili a fantasmi in microscopia ottica, composti da lipidi per il 41%, proteine per il 52% e carboidrati per il 7%.
L’esecuzione di una elettroforesi sulle proteine di membrana dimostra la presenza di proteine periferiche (del reticolo intracellulare) e proteine integrali (transmembrana). La spectrina é un dimero α/β coiled-coil, localizzato subito sotto la membrana dei globuli rossi che possiede una testa che permette l’interazione omofilica diretta ed una coda che media le interazioni omofiliche indirette. La F-actina/Banda 4.1 sono proteine che stabilizzano l’interazione coda-coda fra le spectrine; la glicoforina A è una proteina transmembrana che lega lo scheletro sottocellulare alla membrana dei globuli rossi, determinando la specificità dei Gruppi MN sulla superficie cellulare e l’ankirina è una proteina che media il legame fra le teste di spectrina e la Glicoforina A.
EZIOLOGIA:
Esistono numerose cause di sferocitosi ereditaria, tutte legate a mutazioni enzimatiche che possono portare a riduzione di spectrina, riduzione di proteina 3, mutazioni nella coda di spectrina (nel punto di legame con la Banda 4.1), mutazioni nel dimero, ecc… Tali mutazioni genetiche comportano alterazioni morfologiche nella cellula che diviene sferica, ed alterazioni funzionali con minor plasticità/deformabilità, alterato metabolismo cellulare, maggiore fragilità osmotica, alterata viscosità citosolica e maggiore concentrazione emoglobina. Tali alterazioni funzionali si riflettono in alterazioni morfologiche (con comparsa di sferociti), una ridotta deformabilità nei sinusoidi splenici ed una maggiore fagocitosi macrofagica.

Clinicamente la malattia si manifesta con una triade classica dell’anemia emolitica con sferocitosi periferica, ittero e splenomegalia, anche se con intensità differente. Nella forma lieve (che si ha in un quarto dei pazienti) a parte una lievissima anemia (compensata da una maggiore eritropoiesi) si ha una clinica silente che si aggrava solamente durante gli stati infettivi, lo stress fisico e la gravidanza. Nella forma moderata (che si ha in due terzi dei pazienti) si hanno quadri di emolisi non compensata, periodi intermittenti di ittero (dovuti a fattori scatenanti) e splenomegalia di intensità variabile. Nella forma grave (presente in meno del 10% dei pazienti) si ha anemia grave (richiedente trasfusioni), ritardo di crescita, deformità delle ossa craniche ed ulcere agli arti inferiori.
Complicanze che vanno tenute a mente sono la possibile comparsa di colelitiasi (condizione frequente, tipica di tutte le anemie emolitiche), la crisi emolitica provocata da fattori scatenanti che solitamente non è grave, ma richiede comunque una terapia trasfusionale e la crisi aplastica condizione rara e grave ad esito fatale con febbre, dolore addominale, vomito, anemia e reticolopenia della durata di 10-14 giorni che solitamente è scatenata da una co-infezione di Parvovirus B19.
Al laboratorio si dimostra la presenza di anemia emolitica ed una fragilità osmotica che scompare dopo incubazione con ATP; si pone una corretta diagnostica differenziale con altre forme di anemie immunoemolitiche per la negatività del Test di Coombs. La terapia é di tipo patogenetico: la splenectomia riduce di molto la gravità dell’anemia, anche se è meglio non effettuarla prima dei 5-10 anni d’età.
5) ANEMIE IMMUNOEMOLITICHE:
Le anemie immunoemolitiche sono delle condizioni in cui le emazie sono prematuramente distrutte a causa della loro interazione con auto-anticorpi diretti contro gli antigeni delle emazie. Si classificano in forme da auto-anticorpi se la produzione di anticorpi è diretta contro antigeni presenti sulle proprie emazie ed in questa categoria sono comprese anche le forme indotte da farmaci, oppure in forme da iso-anticorpi se la produzione di anticorpi è diretta contro antigeni presenti sulle emazie ma non di se stessi; un esempio tipico è dovuto alle reazioni da trasfusioni e/o alla gravidanza.
CARATTERI GENERALI:
Per quello che concerne gli antigeni sulle emazie si hanno gli antigeni-polisaccaridici che sono i più resistenti ai processi di laboratorio e più facilmente caratterizzabili chimicamente. Sono gli antigeni chiamati AB0, l’antigene Lewis, il gruppo P, e gli antigeni I/i; sono antigeni a diffusione ubiquitaria (presenti anche nei batteri del tratto gastro-enterico) e verso di essi possono esistere degli anticorpi naturali, così chiamati perché sono presenti verso tali antigeni pur senza una apparente esposizione. Ovviamente tale esposizione é avvenuta e si pensa che questa sia avvenuta durante il contatto con i batteri intestinali che son ricchi di questi antigeni. Gli antigeni-proteici invece sono sostanze chimicamente meno note (perché alterate durante i processi di purificazione) verso cui non esistono anticorpi naturali perché tali antigeni sono degradati già durante la digestione enzimatica e non vengono mai visti dall’organismo. Sono gli antigeni Rh, Kelly e Duffy.
Per quello che concerne gli anticorpi che intervengono in queste reazioni esistono auto-anticorpi diretti contro gli antigeni eritrocitari del self (che portano alla definizione di autoimmunità) e gli iso-anticorpi diretti contro antigeni eritrocitari altrui (che portano a parlare di alloimmunità). Per quello che riguarda la classe immunoglobulinica esistono sia le IgM (a struttura pentamericia, che legano soprattutto gli antigeni polisaccaridici) e le IgG (a struttura monomericia, derivate da IgM dopo lo swith isotipico, che legano soprattutto antigeni proteici ed i farmaci), quest’ultime in grado di attraversare la placenta (generando anemia emolitica del neonato). Gli anticorpi sono caratterizzati da una termicità intesa come temperatura ottimale alla quale avvengono le diverse reazioni immunoemolitiche, solitamente classe-specifici (anche se esiste la possibilità eccezioni): le IgM sono generalmente anticorpi freddi (che sono ben attivi a 4°C, dove si hanno modificazioni conformazionali), mentre le IgG sono anticorpi caldi (ben più attivi a 37°C).
Per quello che concerne la capacità agglutinante la superficie cellulare dei globuli rossi è carica negativamente (con un potenziale elettrico z di circa 15 mV), che determina un reciproco allontanamento fra un globulo rosso ed un altro. Gli anticorpi che sono in grado di ben agglutinare si chiamano anticorpi completi e sono IgM che superano tale forza repulsiva presente fra due globuli rossi, provocando agglutinazione, mentre gli anticorpi incompleti sono IgG che non sono in grado di superare il potenziale elettrico e non possono avvicinare due globuli rossi. Per quello che invece riguarda la capacità del complemento, la reazione antigene-anticorpo attiva la via classica del complemento (C1, C4, C3) che si ha quando esistono due porzioni Fc molto vicine fra loro: a tal riguardo le IgG sono monomeri che attivano il complemento ad alta concentrazione, mentre le IgM sono pentameri che attivano il complemento molto più facilmente, anche se loro sono attive a 4°C ed il complemento a 37°C, per cui solitamente a temperature standard tale attivazione non avviene.

MECCANISMI DI EMOLISI:
L’emolisi si definisce intravascolare quando il complemento si attiva fino ad arrivare al MAC con C8-C9 (che funziona solo se si ha una fissazione adeguata a 37°C) per IgG ad alta concentrazione e/o IgM calde che si fissano sulla parete del globulo rosso. Come conseguenza si ha una perforazione dei globuli rossi con rilascio di emoglobina e composti intracellulari. L’emoglobina inizialmente viene fissata dall’aptoglobina in maniera irreversibile, con rapporto stechiometrico 1:1, per poi essere rimossa dal fegato; quando l’emolisi satura completamente la capacità tamponante dell’emoglobina, questa viene ancora riassorbita a livello renale (tubulo contorto prossimale) e quando anche questo sistema viene saturato, compare ematuria.
L’emolisi si definisce extravascolare invece quando è dovuta ad un incremento della normale emocateresi splenica: le IgG incomplete (o le IgM fredde) lasciano legato ai globuli rossi la porzione C3b (mentre la frazione C3a è solubile), a funzione opsonizzate. Sui macrofagi ricordiamo che esiste il recettore Fc-r diretto contro la porzione Fc delle immunoglobuline ed il recettore C3b-r specifico per il C3b attivato. Il C3b difatti deve scindersi in C3c e C3d con il C3b-inattivatore che rilascia il C3d sulla cellula ed il C3c legato al macrofago. A questo punto si ha l’eliminazione extravascolare dei globuli rossi opsonizzati


TEST DI COOMBS:
Il test di Coombs è un test di emagglutinazione in vitro, dove viene utilizzato del sangue anticoagulato, su cui si esegue un test diretto ed indiretto. Il test diretto valuta la presenza di anticorpi e/o del complemento adesi alle emazie: si utilizzano i globuli rossi del paziente, incubandoli con il siero di Coombs, che altro non é che siero di coniglio diretto contro le IgG umane ed altri anti-sieri, valutando se avviene l’agglutinazione per avere la positività del test. Il test indiretto valuta la presenza di anticorpi liberi nel siero del paziente: si prende il siero del paziente e dei globuli rossi note (chiamate emazie test), cui si aggiunge il siero di Coombs (ed altri antisieri) e si valuta se avviene l’agglutinazione.


5.1) ANEMIE IMMUNOEMOLITICHE DA ANTICORPI INCOMPLETI CALDI
Le anemie emolitica autoimmuni da anticorpi incompleti caldi sono anemie emolitiche caratterizzate da un’emolisi secondaria alla presenza di IgG (raramente da IgA) dirette contro i propri eritrociti. La malattia presenza un’incidenza di 1:80.000 casi annui, con insorgenza in qualsiasi età (soprattutto attorno ai 40 anni); non esiste una predisposizione razziale e vengono prevalentemente colpite le donne. Solitamente gli anticorpi sono diretti contro il fattore D (Rh).
Dal punto di vista eziologico il 40% sono forme idiopatiche, anche se la reale incidenza di malattia è ampiamente sottostimata; altre cause sono le forme autoimmuni (da leucemie, linfomi, mielomi) o più raramente da infezioni. Le forme da farmaci invece possono svilupparsi secondo tre differenti meccanismi: un primo meccanismo é lo stimolo degli auto-anticorpi per cui si induce autoimmunità per la presenza di un fattore estrinseco che poi non prende parte direttamente al processo autoimmune, forse legato all’HLA-B27 e/o alla down-regulation dei linfociti T soppressori. Tale meccanismo é tipico dell’α-metildopa (30-40% dei pazienti a 6 mesi, di cui 1% manifestano malattia clinica), della levodopa, ecc… ed é reversibile alla sospensione del farmaco. Un secondo meccanismo é legato alla funzione aptenica: il farmaco si lega all’antigene dei globuli rossi, stimolando la produzione di auto-anticorpi; solitamente si ha un quadro clinico improvviso e brusco. È un meccanismo tipico della Penicillina ad alte dosi (oltre 20.000 UI die, come nella terapia dell’endocardite) ed é reversibile alla sospensione. Un terzo ed ultimo meccanismo é la formazione di immunocomplessi che si legano sui globuli rossi, generano attivazione del complemento e successivamente si staccano per andare a legare altri globuli rossi; sono immunocomplessi che generano emolisi intravascolare. E’ un meccanismo tipico della Chinidina.
Il meccanismo patogenetico comune é che le IgG ricoprono le emazie, ma non si ha attivazione del complemento (se non ad alte concentrazioni anticorpali); si genera perciò un’emolisi extravascolare per opsonizzazione delle emazie e rimozione a livello della milza e dei sinusoidi epatici.
Il quadro clinico di questi pazienti non è particolare e del tutto sovrapponibile alle altre anemie), ma si può classificare in base all’esordio; le forme insidiose mostrano anemie emolitiche di modesta entità, con subittero e lieve splenomegalia, mentre le forme acute presentano un esordio fulminante (spesso letale), con intensa rigenerazione midollare dei globuli rossi, presenza di punteggiatura basofila e colorazione grigiastra (anziché rosata) e rappresenta la forma severa della malattia Al laboratorio di biochimica si ha un netto aumento della bilirubina indiretta con riduzione dell’aptoglobina, mentre nello striscio di sangue periferico si riscontra anemia, reticolocitosi e sferociti per rimozione di parte della membrana dei globuli rossi da parte dei macrofagi.
Il test di Coombs risulta variabile: per le forme lievi risulta positivo il test diretto per le IgG), nelle forme moderate risulta positivo il test diretto per le IgG, negativo per il complemento D3 e negativo per il test indiretto) mentre nelle forme severe risulta positivo per le IgG e C3, a volte anche per il test indiretto). Il 3% dei pazienti con Coombs negativo ha un numero dianticorpi adesi alle emazie molto basso, inferiore al limite di detettabilità dei kit.
La malattia ha una andamento cronico, con fasi di remissione e riacutizzazione e la sopravvivenza a 5 anni è del 70%, mentre un 20-30% ha un’evoluzione fatale. La terapia prevede l’uso di corticosteroidi che risultano efficaci nel 70% dei casi (addirittura un 20-30% dei pazienti ha una remissione completa), non per immunosoppressione, ma per il blocco della funzione macrofagica. Nel caso di una terapia prolungata si ha anche effetto immunosoppressivo. La splenectomia riduce l’entità dell’emolisi extravascolare nel 50% dei casi, mentre le trasfusioni sono poco efficaci in acuto per l’effetto di emolisi indotta dagli anticorpi. Le immunoglobuline somministrate IV ad alte dosi, hanno funzione di saturare i recettori macrofagici e possono servire per le forme più severe di malattia.
5.2) ANEMIE IMMUNOEMOLITICHE DA ANTICORPI COMPLETI FREDDI:
Le Anemie Emolitiche Autoimmuni da anticorpi completi freddi sono un gruppo di patologie caratterizzate da emolisi secondaria alla formazione di IgM dirette contro i globuli rossi del proprio corpo; prendono anche il nome di crioagglutinine. Gli antigeni verso cui sono diretti fanno parte del sistema I/i, il sistema antigienico eritrocitario coinvolto nel 90% dei casi; “i” è un antigene saccaridico presente sulle emazie alla nascita, che con il tempo si trasforma verso il fenotipo “I” (nel 98% dei pazienti). Mediante delle emazie test (di un paziente adulto e dal cordone ombelicale) si dimostra la presenza di tali anticorpi. Altri sistemi sono coinvolti più raramente, nel qual caso sono gli antigeni H, M ed N.
Dal punto di vista eziologico si possono avere delle forme idiopatiche, anticorpi monoclonali generati da cause imprecisate, solitamente composti da una sola catena leggera (soprattutto la catena k), forse da plasmacellule iperproducenti, con un andamento cronico e tipico del paziente anziano. Altre cause possono essere le malattie proliferative da linfomi, leucemie, mielomi, malattia Waldestrom, ecc… che possono produrre crioagglutinine dirette verso entrambi gli antigeni I/i. Infine le malattie infettive possono portare a crioagglutinine policlonali, solitamente da M. pneumoniae, CMV, Orthomyxoviridae.
Il meccanismo patogenetico vede le IgM che si legano alle emazie a 4°C (mai al di sopra dei 31°C) tipicamente nel periodo invernale, soprattutto nel naso, alle dita, ai lobi delle orecchie ed in tutte le porzioni periferiche del corpo più fredde. Le IgM si distaccano quando aumenta la temperatura. Il complemento si lega a 37°C, ma non funziona al di sotto di 12°C; attorno ai 10°C può legarsi assieme alle IgM, ma funziona solamente a temperature attorno ai 37°C, per cui generalmente non funziona là dove le IgM sono legate. Inoltre esiste un meccanismo di protezione da parte dei fagociti: esiste il C3b-inattivatore, enzima estremamente rapido, che scinde il C3b e lo trasforma in C3d che, da solo, lo protegge dalla fagocitosi ed evita nuovo legame di C3b.
Clinicamente le forme monoclonali hanno un esordio graduale, cronico, che peggiora con la stagione fredda; a volte l’unica manifestazione clinica è il fenomeno di Raynaud. Le forme policlonali invece vedono una produzione più rapida di anticorpi, ma raramente si ha una malattia clinica. In caso di manifestazione si ha pallore, dolori lombari, febbre, epato/splenomegalia, ittero, reticolocitosi e vomito/diarrea. Al laboratorio si ha un test di Coombs che risulta positivo nella forma diretta verso il complemento, negativo quello verso le IgM (dato che si lavora a temperatura ambiente). L’aspetto importante è quello di valutare con le emazie test se sono diretti contro gli antigeni I/i. Si può avere malattia con un alto titolo nelle forme monoclonali (attorno a 1:1.000, 1:256.000), o medio titolo nelle forme policlonali (attorno a 1:32, 1:128). Nel sangue periferico si hanno delle emazie impilate, per cui appare difficile la conta eritrocitaria. Al laboratorio di chimica si ha iperbilirubinemia indiretta, riduzione dell’aptoglobina ed eventuale picco monoclonale IgM.
A livello prognostico il paziente deve evitare l’esposizione al freddo così da evitare ulteriori crisi emolitiche; le forme idiopatiche/linfoproliferative hanno una prognosi variabile, tipicamente legata alla malattia di base. Nelle forme infettive si ha invece tipicamente un’autorisoluzione con il migliorarsi della malattia. La terapia si basa sull’uso di corticosteroidi che risultano efficaci nel 70% dei casi (il 20-30% va incontro a remissione completa), non per immunosoppressione, ma per il blocco della funzione macrofagica; in caso di terapia prolungata si ha anche effetto immunosoppressivo. La splenectomia funziona poco, dato che si ha un’emolisi di tipo intravascolare. La terapia immunosoppressiva funziona, anche se genera gravi effetti collaterali; per quello che concerne la plasmaferesi è efficace nel ridurre la percentuale di anticorpi circolanti; ma clinicamente é poco efficace.
5.3) ANEMIE IMMUNOEMOLITICHE DA EMOLISINE BIFASICHE:
Le anemie emolitiche autoimmuni da emolisine bifasiche sono fra le forme più rare di anemie immunoemolitiche chiamate anche emoglobinuria parossistica a frigore. In questa tipologia di anemia é presente l’emolisina di Donald-Landsteiner, con delle IgG che agiscono a freddo, selettive per il sistema P. Dal punto di vista eziopatogenetico esistono delle forme idiopatiche (rare), forme secondarie alla Lue (sia congenite che acquisite) o da Virus (rosolia, parotite, varicella, influenza, morbillo, ecc…). L’emolisina bifasica agisce a 4°C ed attiva il fissaggio del complemento; non si genera né stasi né emagglutinazione le IgG sono anticorpi incompleti, ma a 37°C generano lisi intravascolare.
Clinicamente si hanno i sintomi acuti di anemia con emolisi intravascolare con brividi, febbre, dolori lombari, vomito, ecc…, che compaiono dopo qualche minuto/ore dall’esposizione al freddo, persistendo fino a 2 ore dopo tale esposizione. Si può associare una modesta splenomegalia ed emoglobinuria (tipicamente persistente per 24 ore, con urine color “lavatura di carne”). Al laboratorio il test di Coombs risulta positivo nel test diretto per il complemento, non per le IgG (che agglutinano a freddo) ed il test diagnostico è la dimostrazione dell’emolisina di Donald-Landsteiner.
La prognosi solitamente è buona nelle forme secondarie (in quanto risulta sufficiente la terapia eziologica), mentre tende a divenire cronica nelle forme idiopatiche. La terapia si basa soprattutto su una terapia d’evitamento di esposizione al freddo.
6) ANEMIA APLASTICA:
L’anemia aplastica è definita come una pancitopenia del sangue periferico, con associata ipocellularità del midollo osseo, legato ad un deficit primitivo/secondario delle cellule staminali ematopoietiche; la distruzione/alterazione delle cellule staminali ematopoietiche porta pertanto ad un impoverimento del tessuto parenchimale midollare. La malattia é limitata alla linea mieloide, dato che i linfociti mantengono la capacità di replicarsi. Dal punto di vista epidemiologico la malattia ha una incidenza di 2-5 milioni di casi annui, soprattutto nei maschi e l’area geografica più colpita è il territorio asiatico.
EZIOPATOGENESI:
Esistono forme acquisite (che sono le più frequenti), di cui l’80% sono forme idiopatiche, e le forme congenite caratterizzate da una predisposizione al deficit midollare, scatenato da ulteriori fattori acquisiti. La forma idiopatica ad eziologia sconosciuta, é una forma in cui esiste un meccanismo autoimmune, responsabile della soppressione dell’emopoiesi. Tale ipotesi é stata confermata tramite l’analisi delle T-cells in coltura con cellule staminali ematopoietiche dove si é dimostrato un blocco dell’emopoiesi, ma il meccanismo preciso rimane ancora sconosciuto. I farmaci in particolare la chemioterapia con un effetto prevedibile e dose-dipendente porta al blocco ed al danno delle cellule in attiva fase di replicazione; esistono anche altri farmaci che possono causare anemia plastica in maniera dose-indipendente (come il Metimazolo). Alcune tossine chimiche come il benzolo, gli insetticidi, ecc…sono mielotossici e possono provocare aplasia del midollo; anche le radiazioni ionizzanti hanno lo stesso effetto, come dimostrato nelle popolazioni di Hiroshima, Nagasaki, nei lavoratori addetti agli impianti nucleari, nei pazienti esposti alla radioterapia terapeutica, ecc…. Alcune infezioni virali (soprattutto HCV, EBV, CMV ed il parvovirus B19), ma anche T. gondii, Brucella spp e M. tuberculosis possono scatenare episodi di anemia plastica. Infine l’anemia di Fanconi è la forma congenita più frequente, associata ad alterazioni funzionali e strutturali di diversi organi, fra cui il midollo osseo.
I meccanismi patogenetici non sono chiari; in passato ci sono state alcune ipotesi su alterazioni locali con deficit nel microambiente midollare, ipotesi che attualmente sembra da scartare, dato che il trapianto di cellule staminali ematopoietiche sembra avere un buon attecchimento, dimostrando la buona funzionalità del microambiente. Attualmente si pensa che possano esistere delle alterazioni esterne all’ambiente midollare, probabilmente legato a fattori esogeni (come l’autoimmunità) che portano ad un danno delle cellule staminali ematopoietiche; nelle forme idiopatiche si pensa esistano alterazioni cellulari che generino un insulto genico alle cellule staminali ematopoietiche, con esposizione di nuovi antigeni ed attivazione delle T-cells che vanno a distruggere tali cellule. All’anatomia patologica il midollo osseo è chiaramente ipocellulato, con molti spazi midollari vuoti popolati solamente da cellule adipose, uno stroma fibroso e qualche focolaio di linfociti; alla biopsia osteo-midollare si può avere un dry-tap (letteralmente «rubinetto asciutto») proprio per l’ipocellularità locale. Sono anche tipiche altre alterazioni cellulari legate alla presenza di infezioni, tossine, farmaci, ecc… con gigantismo cellulare ed elementi di polinuclearità, ecc….
DIAGNOSI:
I sintomi che il paziente manifesta sono legati al grado d’anemia ed alla velocità d’insorgenza dell’anemia stessa. Nelle forme acute i sintomi sono conseguenti alla granulocitopenia con aumento del rischio di infezioni, ulcere cutanee, febbre, ecc… a rapida comparsa (l’emivita dei granulociti é di 1 giorno) e trombocitopenia associata con diatesi emorragica, a comparsa più tardiva (l’emivita dei trombociti é di 8 giorni). Nelle forme croniche i sintomi sono dominati da quadri di anemia con astenia muscolare, pallore, alterazioni cardiovascolari, mentre altri sintomi sono più rari.
Al laboratorio si ha un emocromo con anemia normocromica normocitica, leucopenia con linfocitosi relativa e trombocitopenia; nel sangue periferico non si hanno alterazioni morfologiche particolari ma un aspetto di sangue diluito. Si parla di anemia aplastica grave se si ha una grave ipoplasia midollare, una trombocitopenia inferiore a 20 G/l, polimorfonucleati inferiori a 0,5*G/l e dei reticolociti inferiori all’1%. All’analisi del midollo osseo si ha un aspetto analogo a quello descritto in anatomia patologica; bisogna ricordarsi che esistono falsi negativi se il prelievo cade su piccoli foci di midollo ipercellulato. In questo caso è meglio eseguire (in anestesia generale) altri prelievi per la diagnosi. La diagnosi di certezza la pone l’analisi del midollo, anche se esistono numerose forme cliniche che si pongono in diagnostica differenziale, come la mieloftisi (per alterazioni ematologiche/neoplastiche), l’anemia refrattaria, l’emoglobinuria parossistica notturna e la splenomegalia massiva (di cui si esegue la diagnosi differenziale mediante l’analisi del midollo).

PROGNOSI E TERAPIA:
Le forme secondarie sono forme che regrediscono in poco tempo dopo la rimozione della causa eziologica, generalmente avviene in poche settimane, con andamento benigno. Le forme idiopatiche presentano un decorso variabile (che va dal recupero fino alla morte) in funzione di diversi fattori prognostici negativi (quali il sesso maschile, la manifestazione acuta, la forma emorragica e la forma severa).
La terapia eziologica è efficace nelle forme secondarie. Per le forme primarie sono state studiate diverse terapie, seppure con scarsi risultati. Gli androgeni hanno un effetto stimolante sull’Epo, con maggiore reclutamento delle cellule staminali ematopoietiche e nuova proliferazione (anche se per funzionare adeguatamente richiede la presenza di una popolazione residua di cellule staminali ematopoietiche. L’immunosoppressione si attua mediante globuline anti-linfocitarie/timocitiche e/o immunosoppressori ma risultano efficaci nelle forme primitive di cui bisogna essere sicuri ci sia un’eziologia immuno-mediata. Infine rimane il trapianto di midollo che generalmente si esegue da fratello HLA-aploidentico e che rappresenta la terapia d’elezione nelle anemie aplastiche gravi.
7) EMOGLOBINURIA PAROSSISTICA NOTTURNA:
L’emoglobinuria parossistica notturna è un’anemia emolitica cronica, causata da un deficit acquisito della membrana eritrocitaria; si caratterizza per emolisi intravascolare, maggiore sensibilità alla lisi da parte del complemento ed una presenza di un clone di cellule anomale. La malattia rappresenta l’unico esempio di anemia emolitica con mutazione genica acquisita della membrana cellulare e presenta un’incidenza massima attorno a 20-40 anni d’età, con ugual rapporto maschi/femmine. Non esiste familiarità e/o predisposizione razziale.
La causa eziologica é una mutazione somatica del gene PIG-A sul cromosoma X, nella sua GPI-anchor indispensabile per il legame delle diverse molecole di superficie. Si ha quindi il distacco di CD59 (chiamato anche MIRL, Membrane Inhibitor of Reactive Lysis) e CD55 (DAF, Decay Accelerating Factor) che antagonizzavano il complemento. Il globulo rosso solitamente è protetto dalla lisi del complemento mediante CD55/CD59; l’assenza di tali proteine rende queste cellule più vulnerabili alla lisi del complemento (sia per la via classica che alternativa), con aumento dell’emolisi intravascolare. I quadri clinici possibili sono differenti in funzione della differente percentuale di cellule anomale: le cellule EPN I sono emazie visivamente normali, le EPN II sono emazie che appaiono più sensibili alla lisi di 6-7 volte rispetto alla norma, mentre le EPN di tipo III hanno una sensibilità di 20-25 volte maggiore rispetto alla norma.

FISIOPATOLOGIA:
La malattia si caratterizza per la presenza di crisi intravascolari, crisi che si sviluppano soprattutto la notte, con emoglobinuria (le cellule responsabili sono le cellule EPN III); il 20% dei pazienti non riscontra alcuna emolisi, il 20-50% dei pazienti riscontra una minima emolisi, mentre oltre il 50% dei pazienti manifesta una emoglobinuria costante. Un altro elemento sono le trombosi venose dato che sui leucociti non si lega il recettore dell’urochinasi (u-PAr), importante per la generazione di plasmina, trovandosi quindi in uno stato di diatesi trombofilica. Per quello che concerne le infezioni il paziente presenta una propensione alle infezioni batteriche, soprattutto per la sua leucopenia (spesso generata da una co-esistente anemia aplastica) e per l’alterazione funzionale dei granulociti. Infine, come appena accennato, l’anemia aplastica é un altro fattore, dato che esiste una forte associazione fra tali patologie, per cui dopo anni di anemia aplastica si può sviluppare emoglobinuria parossistica notturna forse perché le cellule staminali ematopoietiche mutate acquisiscono un vantaggio selettivo, divenendo più resistenti alla distruzione immunologica e viceversa.
DIAGNOSI:
A livello clinico l’emoglobinuria é l’elemento fondamentale e si presenta all’esordio in un quarto dei casi (chiaramente compare in tutti i pazienti dato che é l’elemento diagnostico cardine), tipicamente con una circadianità con il ritmo sonno-veglia (soprattutto al mattino), una differente durata (a volte giorni, a volte settimane) e comportante una progressiva anemizzazione con eventuale ittero associato. Le trombosi venose profonde sono la principale causa di morte (nel 50% dei pazienti) che può coinvolgere gli arti inferiori, il sistema portale, il sistema mesenterico (con dolore addominale), portare alla sindrome di Budd-Chiari (30%), ecc… La disfagia può essere un altro segno, alle volte anche estremamente severo, con contrazioni esofagee fino a 10 volte più forti della norma, la cui patogenesi é sconosciuta. Infine le infezioni anche opportuniste sono un altro elemento associato, talmente importanti da essere responsabili del 10% delle morti di questi pazienti.
Fra gli esami di laboratorio all’emocromo si riscontra un’emoglobina inferiore a 6 g/dl, con trombocitopenia, mentre nel sangue periferico si riscontra macrocitosi, reticolocitosi relativa, senza alcuna alterazione nei trombociti. Gli esami biochimici mostrano un netto incremento delle LDH e dei valori sierici di bilirubina (soprattutto la forma indiretta), con riduzione della sideremia (per perdita ematica cronica) con un test di Coombs negativo. Nelle urine si riscontra emoglobinuria ed alla colorazione di Pearl emosideruria.
La diagnosi si pone tramite il Test di Ham dove si utilizzano dei globuli rossi del paziente che vengono posti in un siero di controllo (ricco di elementi del complemento) che si acidifica progressivamente. A pH acido le cellule EPN III vanno incontro a lisi. Ad oggi questo metodo non viene più utilizzato, in quanto vecchio e ricco di falsi positivi. La citofluorimetria invece dimostra una doppia popolazione di cellule EPN positive/negative, dove il citofluorimetro seleziona le diverse cellule in base alla presenza/assenza di CD55/CD59 e valuta la presenza di questa doppia popolazione di cellule.


PROGNOSI E TERAPIA:
La malattia é cronica e presenta una sopravvivenza media di 10 anni; la gravità e la prognosi dipendono da tanti altri fattori, quali la percentuale di cellule EPN II/III, il grado di ipoplasia midollare e la frequenza di infezioni/trombosi venose. Nel 3% dei pazienti si può avere una remissione totale, mentre il 50% dei pazienti mostra un miglioramento con il tempo; raramente si ha lo sviluppo di leucemia mieloide acuta (la malattia viene difatti considerata una sindrome mielodisplastica dei globuli rossi). La terapia é principalmente di supporto con implementazione di ferro, acido folico ed Epo ricombinante, mentre la somministrazione di androgeni o cortisonici sono poco efficaci (qualche studio ha documentato qualche minimo effetto dei cortisonici nelle crisi acute, ma la risposta non é chiara). La terapia risolutiva appare quella di eseguire il trapianto di cellule staminali ematopoietiche.
REFERENCES:
: Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001;345:1368-1377
: Nutritional deficiencies and blunted erythropoietin response as causes of the anemia of critical illness. J Crit Care 2001;16:36-41
: Erythropoietin gene expression is suppressed after lipopolysaccharide or interleukin-1 beta injections in rats. Am J Physiol. 273:t-71 1997
: Proinflammatory cytokines lowering erythropoietin production. J Interferon Cytokine Res 1998;18:555-559
: Progress in understanding the pathogenesis of the anemia of chronic disease. Blood 1992;80:1639-1647
: Pathogenesis and treatment of the anemia of chronic disease. Am J Med Sci 1994;307:353-359
: High-dose recombinant human erythropoietin stimulates reticulocyte production in patients with multiple organ dysfunction syndrome. J Trauma 1998;44:361-367
Efficacy of recombinant human erythropoietin in the critically ill patient: A randomized, double-blind, placebo-controlled trial. Crit Care Med. 1999;27:2346-2350
: Efficacy of recombinant human erythropoietin in critically ill patients: A randomized controlled trial. JAMA22002;88:2827-2835
: EPO Critical Care Trials Group. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med2007;357 (10):965-976
: Erythropoietin-receptor agonists in critically ill patients: a meta-analysis of randomized controlled trials.CMAJ 2007;177 (7):725-734
12.: A randomized double blind placebo controlled trial of recombinant human erythropoietin in long term acute care patients. Crit Care Med 2003;31:A167
: Anemia of the critically ill: “Acute” anemia of chronic disease.Crit Care Med 2000;28:3098-3099


0 Response to "Anemia in ICU - forme specifiche (Capitolo 2.10b.1-2)"
Posting Komentar