

L’emoglobina è la proteina più rappresentata nei globuli rossi ed ha la funzione principale di trasportare l’ossigeno proveniente dall’esterno ai diversi tessuti, veicolando l’anidride carbonica in senso opposto. Ne parleremo della sua importanza dal punto di vista fisiopatologico sia nella reologia emodinamica che a livello respiratorio nei capitoli dedicati all’ipossia ed agli scambi gassosi (sezione 3). In questa prima parte iniziamo ad introdurre dei concetti di base della produzione dell'emoglobina e dell'anemia in ICU, per poi concentrarci meglio su alcune anemie importanti da conoscere in Medicina Intensiva (si veda il prossimo capitolo, Capitolo 2.10b.1-2) ed infine la terapia sostitutiva in caso di anemia (Capitolo 2.10b.2).
Lo sviluppo dell’emoglobina inizia con il riscontro di due cluster globinici chiamati cluster α-globinico che é mappato sul cromosoma 16 e si compone delle sequenze ξ (zaeta), ψ (psi) ed α (alfa) ed il cluster β-globinico che é mappato sul cromosoma 11 composto a sua volta dalle sequenze ε (epsilon), γ (gamma), δ (delta) e β (beta). A monte di tali cluster si hanno delle sezioni chiamate LCR (Locus Control Region) sensibili alle DNAasi, che controllano in Cis le diverse sequenze appena indicate e la loro differente espressione, con una correlazione spazio-temporale.


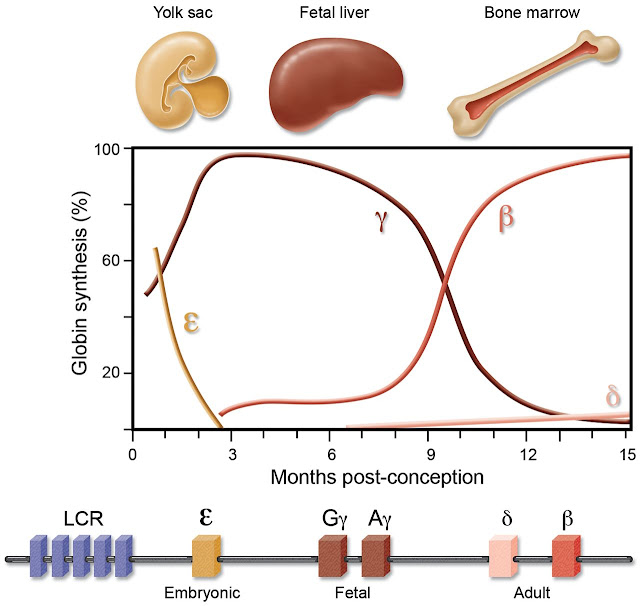
Vuol dire che il controllo in Cis avviene nei primi periodi di tempo nelle regioni più vicine alle LCR e con il passare del tempo si ha anche un cambiare lo spazio di stimolazione; nell’embrione l’emoglobina compare durante la VI-X settimana di gestazione: si ha la comparsa dell’Emoglobina Gower 1 (composta dalle catene ζ2, ε2), seguita dall’Emoglobina Portland (catene ζ2, γ2) ed infine l’Emoglobina Gower 2 (composta dalle catene α2, ε2). Nel feto viene espressa l’Emoglobina Fetale (chiamata HbF e composta dalle catene α2, γ2), che rappresenta l’emoglobina più frequente e che predomina durante l’intera vita fetale; alla nascita è ancora il 60% di tutta l’emoglobina circolante, per poi raggiungere i valori “adulti” attorno ad 1 anno. Nall’adulto infine si hanno l’emoglobina A1 (HbA) composta dalle catene α2, β2 che rappresenta il 97% dell’emoglobina totale, l’emoglobina A2 (HbA2) composta dalle catene α2, δ2 che rappresenta il 2-3% dell’emoglobina totale ed in minima parte l’emoglobina fetale (HbF) composta dalle catene α2, γ2 che normalmente rappresenta lo 0-1% dell’emoglobina totale.



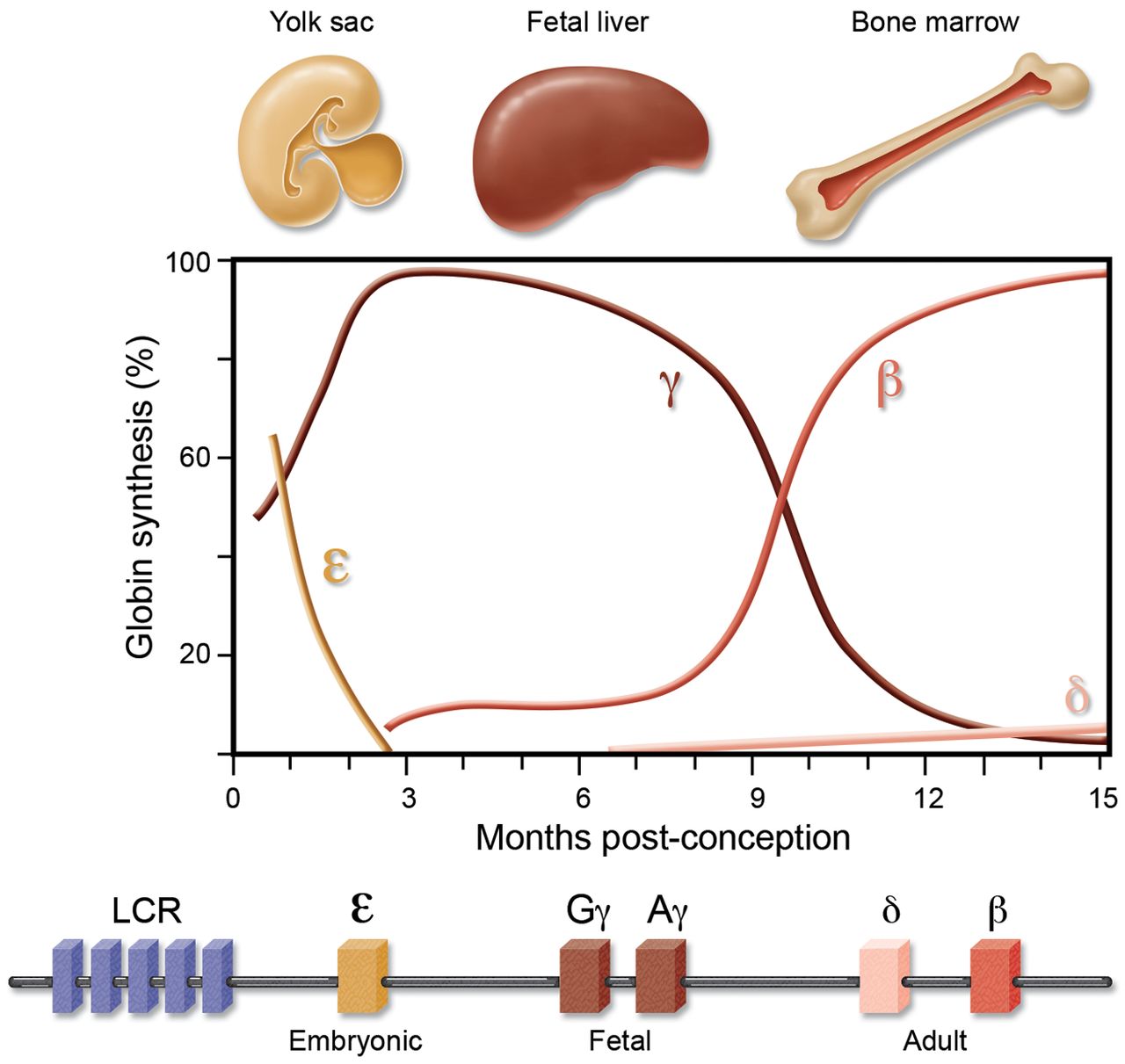
Vuol dire che il controllo in Cis avviene nei primi periodi di tempo nelle regioni più vicine alle LCR e con il passare del tempo si ha anche un cambiare lo spazio di stimolazione; nell’embrione l’emoglobina compare durante la VI-X settimana di gestazione: si ha la comparsa dell’Emoglobina Gower 1 (composta dalle catene ζ2, ε2), seguita dall’Emoglobina Portland (catene ζ2, γ2) ed infine l’Emoglobina Gower 2 (composta dalle catene α2, ε2). Nel feto viene espressa l’Emoglobina Fetale (chiamata HbF e composta dalle catene α2, γ2), che rappresenta l’emoglobina più frequente e che predomina durante l’intera vita fetale; alla nascita è ancora il 60% di tutta l’emoglobina circolante, per poi raggiungere i valori “adulti” attorno ad 1 anno. Nall’adulto infine si hanno l’emoglobina A1 (HbA) composta dalle catene α2, β2 che rappresenta il 97% dell’emoglobina totale, l’emoglobina A2 (HbA2) composta dalle catene α2, δ2 che rappresenta il 2-3% dell’emoglobina totale ed in minima parte l’emoglobina fetale (HbF) composta dalle catene α2, γ2 che normalmente rappresenta lo 0-1% dell’emoglobina totale.

SINTESI DELL’EMOGLOBINA
Ogni globulo rosso contiene circa 640 milioni di molecole di emoglobina, nel rapporto che esiste fra le tre principali isoforme dell’adulto come appena elencato; la sintesi dell’eme avviene nei mitocondri e nel citosol (secondo la via delle porfirine) e contiene un atomo di Ferro legato alla Protoporfirina IX. Si ottiene pertanto un tetrametro con due subunità α e due subunità β (da cui la definizione α2, β2); le singole catene non sono solubili e formano precipitati inttracellulari (chiamati corpi di Heinz), mentre il tetrametro con le molecole mantenute assieme fra loro è solubile. L’emoglobina può subire delle modificazioni conformazionali in base alla differente saturazione d’ossigeno; ad alta concentrazione di ossigeno si hanno catene più vicine fra loro e stabili, mentre a bassa concentrazione di ossigeno si hanno catene che si distanziano fra loro, più lontane, che sono stabilizzate dal 2,3-DPG che viene posto al loro interno.


L’emoglobina ha il compito principale di legare ossigeno a tensioni d’ossigeno di 100 mmHg e rilasciare ossigeno a tensioni pressorie di 40 mmHg; ha anche la funzione di legare l’anidride carbonica a basse tensioni di ossigeno e rilasciare anidride carbonica ad elevate tensioni d’ossigeno. La curva di dissociazione dell’emoglobina è una curva a forma sigmoide, dove esiste una cooperatività positiva fra le diverse molecole (effetto che non esiste nella mioglobina a catena singola) e la possibilità di modificazioni delle molecole da parte dell’ambiente esterno (chiamato effetto Bohr). L’effetto Bohr viene indicato come la tendenza della curva di dissociazione emoglobinica a spostarsi verso destra (cioè facilitare il rilascio di ossigeno) con del incremento 2,3-DPG, riduzione del pH locale, incremento della temperatura ed incremento della CO2. Lo scopo filologico é quello di cedere più facilmente ossigeno all’ambiente circostante quando sono presenti elementi biochimici sfavorevoli e che indicano una difficoltà metabolica del tessuto circostante.
Il catabolismo dell’emoglobina è caratterizzato da una rimozione extravascolare da parte dei macrofagi posti nei sinusoidi splenici; la cellula viene scissa nei diversi componenti, che poi saranno riciclati dall’organismo: la globina viene scissa in aminoacidi (e successivamente utilizzata nel fegato), il ferro si lega alla transferrina e successivamente utilizzata nel midollo e la protoporfirina viene trasformata in bilirubina ed anidride carbonica (e riutilizzata nel fegato).
ERITROPOIETINA:
L’eritropoietina ricombinante umana (chiamata rHuEpo) viene utilizzata in diverse situazioni, soprattutto quando si ha anemia con valori di emoglobina inferiori a 10 mg/dl; esistono diverse formulazioni, come l’Epo α [Eprex, Globuren], l’Epo β [Neorecormon EV] e la Darbopoietina [Aranesp, Nespo], quest’ultima a lento rilascio, 1 volta/settimana, che vengono somministrate al paziente per aumentare la produzione midollare di emoglobina. Per la posologia esistono diversi protocolli di dosaggio, in base alle diverse situazioni cliniche in cui il paziente viene a trovarsi; se la terapia vien utilizzata, si somministra fino al raggiungimento di valori di emoglobina a 12 mg/dl (controllando e sostituendo anche lo status marziale). Da sapere che esistono anche dei pazienti considerati non-responders, dove ad 8 settimane dalla terapia non si ha incremento di emoglobina oltre ad 1 mg/dl rispetto al valore iniziale. Il meccanismo non é chiaro. Gli effetti collaterali sono sintomi simil-influenzali, ipertensione arteriosa (soprattutto nei pazienti dializzati), cefalea, convulsioni ed un aumentato rischio di trombosi (per incremento dell’ematocrito).
ANEMIE IN ICU:
Per Anemia si intende una diminuzione del patrimonio emoglobinico totale dell’organismo al di sotto della soglia di normalità (il cui limite varia con l’età ed il sesso del paziente); per porre tale diagnosi non è quindi sufficiente valutare il numero di red cells, ma il valore totale dell’emoglobina. L’emoglobina normale nel maschio è di 13,5-17,5 g/dl, nella femmina è di 12,5-15,5 g/dl; generalmente si parla di anemia quando si hanno valori inferiori nel maschio di 13,0 g/dl, nella femmina di 12,0 g/dl (11,0 g/dl in gravidanza). L'anemia è un problema comune nei pazienti critici ricoverati in ICU. In effetti, in un recente studio trasversale, il 29% dei pazienti ricoverati mostrava una concentrazione di emoglobina al di sotto dei valori normali ed il 37% di questi aveva bisogno di trasfusioni di emazie.
CLASSIFICAZIONE:
La classificazione morfologica è una classificazione che si basa sulla valutazione delle dimensioni eritrocitarie (misurate in fl - femtolitri), calcolate sulla base dell’MCV (Mean Cell Volume). Si parla di anemia normocitica se l’MCV é attorno a 80-100 fl e solitamente é dovuto ad una patologia cronica (ad esempio la sferocitosi ereditaria, l’emoglobinuria parossistica notturna, alcune emoglobinopatie, la mielosostituzione, l’insufficienza renale, ecc…); si parla invece di anemia microcitica quando l’MCV é inferiore a 80 fl, tipicamente dovuto ad anemia sideropenica, anemia sideroblastica, talassemie, carenza di rame. Si parla invece di anemia macrocitica quando l’MCV é superiore a 100 fl e tipicamente é da ricondurre a mielodisplasia, epatopatia, alcolismo, chemioterapia, anemia aplastica, ipotiroidismo; infine l’anemia megaloblastica si caratterizza per un MCV superiore a 110 flgenerato da mielodisplasia, carenza di vitamina B12/folati, chemioterapia, anemia aplastica, ecc…
Un’altra classificazione é di tipo fisiopatologico e si basa sul dato fisiologico che la massa eritrocitaria (M) è direttamente proporzionale all’emoglobina prodotta dal midollo (H) ed all’emivita delle emazie (t½). Quindi M = H * t½ Si parla pertanto di anemia con riduzione di produzione per cause ipoproliferative (sideropenia, malattie cronice), deficit di Epo (insufficienza renale), ipoplasia midollare (come nel caso di anemia aplastica) o eritropoiesi inefficace (come nel caso della talassemia, ecc…), oppure per riduzione dell’emivita dei globuli rossi come in caso di emorragia o ipercatabolismo (tipico é il caso delle anemie emolitiche). Infine esistono altre forme patologiche di anemia in base a tale classificazione fisiopatologica, che sono dovute ad emodiluizione (in particolare la gravidanza, la splenomegalia ed una idratazione eccessiva).
FISIOPATOLOGIA DELL’ANEMIA:
Le modificazioni nelle concentrazioni plasmatiche di emoglobina provocano delle alterazioni nei sistemi corporei a diversi livelli, che sono a livello cardiovascolare dove si ha un incremento della frequenza cardiaca e dello stroke volume per incrementare la gittata cardiaca (é un meccanismo compensatorio che si instaura in pochi minuti), a livello biochimico, dove si ha un incremento del 2,3-DPG intracellulare, efficace soprattutto a basse tensioni di ossigeno e che lega competitivamente l’emoglobina ossigenata e ne modula l’affinità (mediante l’effetto Bohr), cercando di compensare il deficit periferico con una maggiore facilità di rilascio d’ossigeno ai tessuti (è un meccanismo di compenso che avviene nel giro di 10 ore circa) ed infine a livello emopoietico, dove si ha un incremento renale della sintesi di Epo, con una conseguente stimolazione della eritropoiesi ed incremento dei reticolociti circolanti (é un meccanismo di compenso che si instaura nel giro di diverse ore).
Diventa pertanto importante conoscere anchela velocità d’insorgenza dell’anemia, dato che nelle forme acute si ha solamente l’attivazione del solo sistema cardiovascolare (e per questo generalmente sono sintomatiche), mentre le forme croniche sono lentamente compensate da tutti i sistemi descritti, quindi generalmente sono poco sintomatiche.
CLINICA:
La sintomatologia del paziente anemico dipende da diversi fattori, quali la velocità d’insorgenza della malattia (anemie ad insorgenza rapida sono più sintomatiche per la presenza del solo adattamento cardiaco), la severità dell’anemia (forme moderate a 9-10 g/dl sono meno sintomatiche delle forme gravi, inferiori a 6 g/dl), l’età del paziente (i pazienti anziani tollerano peggio l’anemia perché spesso non esiste il compenso cardiovascolare come nel giovane) ed il metabolismo eritrocitario (per cui pazienti con deficit enzimatici, soprattutto eritrocitari, sono più sintomatici dei pazienti senza tale co-patologia, a parità di deficit di emoglobina). Possono esistere sintomi neuromuscolari come astenia (frequente e riconducibile ad ipoperfusione muscolare), cefalea, vertigini, tinnito e claudicatio intermittens, sintomi cutanei come fragilità ungueale, pallore (soprattutto al palmo delle mani, al padiglione auricolare, alla mucosa delle labbra/congiuntive), telogen effluvium ed eventualmente ittero (da emolisi), sintomi cardio-respiratori come dispnea da sforzo, tachicardia/tachipnea, soffio sistolico da eiezione o angina (quest’ultima in particolare negli anziani) e sintomi gastrointestinali come la stomatite angolare, le papille atrofiche e la disfagia.
ANEMIE MEGALOBLASTICHE:
Le anemie megaloblastiche sono quel gruppo di anemie macrocitiche con eritroblasti anomali (megaloblasti) e macrovalociti posti nel sangue periferico. L’eziologia può essere ricondotta alla vitamina B12 oppure ai volati. Per quello che riguarda la vitamina B12 si può avere un deficit di vitamina B12 che comunque appare essere una condizione molto rara, dato il fabbisogno giornaliero esiguo da parte del corpo, l’entità delle scorte corporee e la ridotta eliminazione. Dovrebbe esserci un deficit di assorbimento per circa 2 anni per avere questa causa. Più spesso si può avere un malassorbimento di vitamina B12 sia da mutazioni al fattore intrinseco (come in caso di anemia perniciosa o gastrectomia), che da malattie intestinali (come in corso di celiachia, ileite, malattia di Chron, ecc…) oppure per consumo della vitamina B12 da parte dei saprofiti intestinali. Infine sono da ricordare altre patologie della vitamina B12 come la mutazione delle transcobalamine, la mutazione del fattore intrinseco o la sindrome di Immerland-Grasbeck (con malassorbimento congenito della vitamina B12). Per quello che invece concerne i filati, si può avere un deficit di folati soprattutto in pazienti anziani, alcolisti ed in tutta la popolazione con una dieta priva di frutta e verdura (questa causa rappresenta la condizione eziopatogenetica più frequente), oppure per un malassorbimento di folati da malattie intestinali o anticonvulsivanti (soprattutto barbiturici) oppure per un maggiore fabbisogno di folati che raggruppa in sé una serie di condizioni estremamente varie, legate alla presenza di una rapida iperproliferazione corporea, come la gravidanza/allattamento, le anemie emolitiche, le infiammazioni croniche, il periodo dello sviluppo, le malattie metaboliche e/o le neoplasie.
FISIOLOGIA VITAMINA B12-FOLATI:
La Vitamina B12 (o cobalamina) ed i Folati sono dei composti presenti in natura (sia nel regno animale che vegetale) che l’uomo assume con la dieta per poter svolgere diverse funzioni cellulari; un deficit importante di queste sostanze provoca anemie megaloblastiche, che si differenziano dalle anemie macrocitiche perché si associa megaloblastosi midollare, un indice importante di malnutrizione, caratterizzata da maturazione nucleare che appare ritardata rispetto allo sviluppo del citoplasma cellulare.
La vitamina B12 é formata da un nucleotide e da un anello corrinico (come le porfirie), contenente al centro un atomo di cobalto; esistono due co-enzimi che sono in grado di utilizzarla: un enzima epatico (deossi-adenosil-cobalamina) ed un enzima plasmatico/cellulare (metil-cobalamina). La vitamina è diffusa negli alimenti di origine animale ed è resistente alla cottura; l’assunzione quotidiana è di circa 5-30 uγ (con un fabbisogno giornaliero di circa 1 γ). Il fattore intrinseco è una glicoproteina costituita da due catene polipeptidiche, prodotte dalle cellule parietali del corpo/fondo gastrico che lega avidamente la vitamina B12 e ne permette l’assorbimento ileale (tramite opportuni trasportatori); dall’orletto a spazzola poi la Vitamina B12 viene staccata e trasportata nel plasma verso le transcobalamine. Di queste transcobalamine ne esistono tre isoforme: le isoforme I e III sono frazioni differenti della cobalofillina (glicoproteine dei granulociti) che legano la vitamina B12 e funzionano come deposito della vitamina; hanno un’emivita di 9-10 giorni. L’isoforma II é una proteina (non una glicoproteine) del peso di circa 30 kDa, che lega debolmente la vitamina B12, agendo come veicolo di trasporto della vitamina stessa; ha un’emivita di circa un giorno.

L’acido folico si compone di acido glutammico, acido paraminobenzoico e pteridina; tale struttura può trovarsi in grosse quantità nei poliglutammati (come i folati, che sono un gruppo eterogeneo di molecole). Generalmente si trova nei vegetali, nel fegato crudo, nel lievito, ecc… ed il fabbisogno quotidiano è di circa 100-200 uγ, con un deposito epatico di 5-10 mg (circa il 50% dell’organismo) e le concentrazioni plasmatiche attorno ai 10 γ/l. La sua funzione é principalmente di intervenire nella biosintesi delle purine, nella biosintesi delle pirimidine e nelle trasformazioni aminoacidiche (la Serina in Glicina, l’istidina in acido glutammico, l’omocitesina in Metionina).
Dal punto di vista della fisiologia, la sintesi di timidilato è uno step necessario per la produzione di DNA; si parte dall’Uridina (che poi diviene deossi-uridina, deossi-uridilato, poi timidilato) per arrivare alla sintesi del DNA. Nell’ultimo step agisce un enzima chiamato timidilato sintetasi, che trasforma la 5,10-metilen-tetraidrofolato in di-idro-folato. Fra il tetraidrofolato ed il 5,10-metilen-tetraidrofolato si ha la conversione di Serina in Glicina, così da acquisire un Metile, che può facilmente donare al Deossi-uridilato. L’altro step importante é l’ingresso del folato nella cellula: il deposito dei folati è il 5-metilen-tetraidrofolato che riesce ad entrare nella cellula grazie alla vitamina B12 (con un rapporto fra siero:globuli rossi di 1:30). Il 5-metilen-tetraidrofolato diviene tetraidrofolato donando un metile (gruppo –CH3) alla omocisteina (che quindi diviene metionina) ed il trasporto é mediato dalla vitamina B12. In caso di deficit di vitamina B12 si ha un “intrappolamento” di 5-metil-tetraidrofolato e si ha una deficit funzionale di folati.
FISIOPATOLOGIA:
Con un deficit nel metabolismo folati-vitamina B12 la sintesi del DNA è rallentata per un allungamento della Fase S, e la mitosi procede con difficoltà; si hanno cellule midollari con un DNA che si arrestano fra la fase diploide e tetraploide. I megaloblasti sono cellule con un grosso nucleo e citosol: nella fase S aumentano i composti citosolici (fra cui l’emoglobina) ed un deficit di sintesi di DNA rallenta la maturazione nucleare rispetto all’evoluzione della cellula; il nucleo è ancora grosso, con un reticolo cromatinico fine e lasso. Si hanno forme da eritropoiesi inefficace. Tali alterazioni cellulari sono diffuse: tutte le cellule dell’organismo sono colpite, soprattutto le cellule ematiche (per l’alto tasso di replicazione cellulare); si ha quindi la comparsa di granulocitopenia e trombocitopenia; in alcuni casi può anche comparire atrofia mucosale diffusa. Le alterazioni mieliniche sono dovute ad un deficit di vitamina B12: senza un corretto metabolismo della sintesi del DNA si ha un accumulo di metaboliti intermedi che diventano tossici per le cellule nervose (come l’omocisteina), legandosi alla mielina e portandola a degenerazione. In casi gravi si possono avere alterazioni nel cordone spinale posteriore.
DIAGNOSI:
Per quello che concerne la sintomatologia il paziente presenta la classica sintomatologia dell’anemia (vedi sopra), cui si associano alterazioni neurologiche (come il segno di Romberg positivo, le parestesie agli arti inferiori, i deficit muscolari, i disturbi visivi, eventuali patologie psichiatriche, ecc…). All’esame obiettivo si hanno alterazioni cutanee con una cute pallida e gialla, come “cera vecchia”, alterazioni cardiovascolari con incremento del rischio di infarto miocardico acuto, patologie cerebrovascolari, ecc…, alterazioni linguali con una lingua rossa e lucida che perde l’aspetto vellutato per una progressiva iporigenerazione (prende il nome di Glossite di Hunter) ed eventuale epatosplenomegalia (nei casi veramente severi) per aumento del catabolismo cellulare.
Agli esami ematochimici si ha un emocromo con MCV superiore ai 110 fl, riduzione dell’ematocrito ed aumento dell MCH. Allo striscio periferico si ha macrocitosi, anisopoichilocitosi, ovalocitosi (con globuli rossi come “uova di Pasqua”), policromasia diffusa (con punteggiatura basofila), polimorfonucleati ipersegmentati ed un incrementato numero di metamielociti, neutropenia, trombocitopenia ed iporeticolociti. Nel midollo osseo infine si può riscontrare un’iperplasia della serie eritroide, con un incremento degli elementi immaturi (si parla di midollo blu per la numerosa presenza di elementi a citosol basofilo), con megaloblasti in diverse fasi maturative (caratterizzati da un nucleo basofilo, policromatofilo, ortocromatico, ecc…) con eventuale frammentazione nucleare. Altri segni indiretti sono un incremento della bilirubina indiretta (come conseguenza della eritropoiesi inefficace), un incremento delle LDH, una riduzione del dosaggio plasmatico di vitamina B12 e dei folati ed il riscontro (in caso di anemia perniciosa) di anticorpi anti cellule parietali/fattore intrinseco.

La diagnosi si pone mediante la clinica (con un adeguato sospetto diagnostico) e gli esami di laboratorio in grado di confermare tale condizione. Spesso si aggiunge la diagnosi ex juvantibus come “prova terapeutica” con concentrazioni di vitamina a basse dosi (1 μg die di vitamina B12, 100 μg die di folati), controllando si ha una risposta positiva a 7 giorni, sia in termini clinici che di laboratorio. In ICU generalmente si procede con una terapia sostitutiva completa, dati i rischi virtualmente assenti di somministrare tale terapia. La prognosi è buona, soprattutto se si attua una terapia eziologica efficace e si cerca di restaurare i depositi corporei di Vitamina B12 e folati. La terapia è di tipo sostitutivo, con lo scopo di ripristinare i diversi deficit di micronutrienti.
ANEMIA SIDEROPENICA:
L’anemia sideropenica è un’anemia legata al deficit di ferro nell’organismo, che si manifesta come tale quando il ferro è insufficiente per un’adeguata sintesi di emoglobina. Dal punto di vista epidemiologico, tale anemia rappresenta la causa più frequente nel mondo di anemia (3% dei maschi, 20% delle donne) e l’età di massima insorgenza di malattia sia ha fra i 6-20 mesi di vita.
EZIOPATOGENESI:
Il ridotto apporto di ferro è una causa rara ed isolata, che avviene soprattutto in pazienti vegetariani, dato che il Ferro viene chelato da sostanze che ne riducono l’assorbimento (come i nitrati, i fosfati, ecc…). Generalmente un deficit di ferro é più facilmente riconducibile ad un maggiore fabbisogno, condizione a sua volta dovuta a tre diverse condizioni: per l’infanzia bisogna sapere che nel primo anno di vita il bambino triplica il proprio peso e necessita ferro per produrre emoglobina (che si trova soprattutto nei cereali e nella carne); nella gravidanza la donna perde 680 g di ferro (corrispondente a circa un terzo delle proprie scorte corporee) per la produzione di nuovi globuli rossi, per il feto e durante il parto; infine nell’allattamento si perdono 0,5-1 mg die di ferro. Un ridotto assorbimento di ferro é invece una causa rara, che si ha soprattutto in caso di achilia (come per una gastrite atrofica e/o post-resezione gastrica) o da alterazioni intestinali (soprattutto nel tratto Digiuno). La perdita protratta di ferro è la condizione più frequente di deficit marziale, soprattutto per perdita ematica nel tubo gastro-enterico (sia nell’uomo che nella donna) e con le mestruazioni; l’emorragie possono essere di diversa entità, ma bastano 10-15 ml die cronicamente per portare ad un’anemia sideropenica importante.
FISIOLOGIA DEL FERRO:
Il ferro è uno fra gli elementi più rappresentati nell’organismo, l’unico che viene controllato nel suo ingresso (a livello intestinale) e non nell’entità delle perdite (a livello renale). L’apporto di ferro avviene tramite gli alimenti, ogni giorno di 10-20 mg (anche se solitamente se ne assume il 10-20%, con un apporto di 1 mg die). Ad influenzare l’assorbimento differente di ferro intervengono la tipologia dell’alimento e l’assorbimento mucoso. Per la tipologia d’alimento l’emoglobina, la mioglobina e le proteine trasportano il ferro come Eme molecolare, che viene liberato solamente negli Enterociti. Il ferro ionizzato deve essere nello stato ferroso, condizione stimolata dall’acidità gastrica ed acidi (come la vitamina C, la cisteina, il fruttosio, ecc…). Nell’acidità il ferro ferroso lega le proteine con mucopolisaccaridi e/o a basso peso molecolare, mentre in ambiente alcalino lega proteine ad alto peso molecolare, precipitando. A questo punto, interviene l’altro aspetto legato all’assorbimento mucoso: gli enterociti secernono protransferrina, molecola legante il ferro, che lo assorbe in un complesso all’interno degli enterociti. Da qua ha diversi destini: viene liberato in circolo (legato alla transferrina), viene depositato nella cellula (con la ferritina) oppure viene donato ai macrofagi locali (per poi essere eliminato). È negli enterociti che si ha il controllo dell’omeostasi fisiologica, anche se non è chiaro il meccanismo base con cui questo avviene.
IL FERRO NEL CORPO:
La transferrina é una glicoproteina sintetizzata dal fegato, che veicola il ferro nel plasma; esistono diverse isoforme della molecola (come l’aptotransferrina se é priva di ferro, in forma monoferrica se lega un atomo di ferro, in forma diferrica se lega due atomi di ferro). È una proteina in grado di interagire con i recettori transmembrana presenti su molte cellule, per poter immagazzinare il Ferro ferroso nell’organismo; solitamente si ha una maggiore affinità per la transferrina diferrica.

Per quello che invece concerne il deposito di ferro, la ferritina è una grossa proteina di 440 kDa che si dispone a ricoprire un sale ferrico insolubile; ciascuna molecola è in grado di ricoprire il 30% del ferro del proprio peso. Le sue concentrazioni plasmatiche riflettono le concentrazioni intracellulari del ferro presente nell’organismo; da non dimenticare che é anche una proteina di fase acuta che aumenta in corso di infiammazione corporea. L’emosiderina invece è un insieme di ferritina, lipidi, proteine, acido sialico, porfirine, ecc… che serve per deposito del ferro intracellulare, anche se la sua reale funzione non è ancora stata completamente chiarita.
FISIOPATOLOGIA:
Con la progressiva perdita di ferro, nel corpo si instaurano delle situazioni che qui vengono classificate ma che rappresentano un continuum clinico. Inizialmente si ha uno stato di sideropenia prelatente dove si ha una riduzione del ferro a livello del sistema reticolo-endoteliale, con una riduzione dell’Emosiderina; l’unica alterazioni visibile è un incremento dell’assorbimento di ferro senza alcuna alterazione a livello biochimico. In seguito, con l’avanzare del deficit, si sviluppa una sideropenia latente caratterizzata da alterazioni laboratoristiche (con riduzione delle concentrazioni intracellulari di emoglobina, incremento della transferrina, riduzione della ferritina, ecc…) non accompagnato da anomalie ematologiche. Infine si arriva alla vera e propria anemia sideropenica, che rappresenta uno stadio tardivo della sideropenia, inizialmente modesta, ma progressivamente più evidente.
DIAGNOSI:
La clinica é riconducibile ad una sintomatologia aspecifica tipica dell’anemia, cui si associano segni specifici di sideropenia, soprattutto con alterazioni cutaneo-mucose. Si può avere cute secca/anelastica, capelli sottili, fragili e radi, coilonichia (unghie concave) e/o cheilite angolare. È sempre estremamente importante ricercare le cause che provocano l’anemia sideropenica, soprattutto per quello che riguarda il sanguinamento (mono/metrorragie, sangue occulto fecale, endoscopia, ematomi), ma anche per quello che riguarda il malassorbimento, un’eventuale presenza di auto-anticorpi, l’emoglobulinuria parossistica notturna, ecc…
Le analisi di laboratorio mostrano un emocromo con anemia microcitica ipocromica (quindi con riduzione dei valori di MCV, MCH, MCHC) ed eventuale anisopoichilocitosi (riconducibile ad una fragilità cellulare); il midollo osseo alla colorazione di Pearls per identificare il ferro si ha un colore blu (per la carenza marziale). Al laboratorio di chimica si ha una riduzione del ferro sierico, con incremento della transferrina ed una riduzione della ferritina (in assenza di infiammazione).

La patologia si pone in diagnostica differenziale con altre forme di anemia come l’anemia sideroblastica e β-talassemia, dove però le concentrazioni di ferro sono normali o elevate, eventualmente associate ad eritroblasti ad anello, ma anche con condizioni infiammatorie croniche (dove si ha un incremento degli indici di flogosi, ferritina compresa).
PROGNOSI E TERAPIA:
La prognosi dipende dalla malattia di base, ma generalmente è buona, soprattutto se viene trattata con successo la causa. La terapia viene differenziata sulla base della severità e dell’eziologia della malattia: il goal terapeutico è quello di correggere l’anemia e ristabilire i depositi marziali nell’organismo. Pertanto si instaura sia una terapia eziologica volta a trattare la causa di base, sia una terapia fisiopatologica atta a correggere il deficit di ferro (generalmente per via enterale); il fallimento terapeutico è da ricercarsi in un’emorragia persistente, in una mancata compliance terapeutica, in una diagnosi errata, nella presenza di un deficit misto e/o in un malassorbimento del ferro.
Il ferro può essere somministrato per via enterale o parenterale. Nella via enterale si preferisce per pazienti asintomatici o con una lieve anemia: si utilizzano compresse di ferro a pasto a digiuno (per avere un pH gastrico acido) per almeno 6 mesi, con attese di incremento emoglobinico di circa 0.5-1 mg alla settimana; i reticolociti incrementano già a 4-5 giorni, mentre l’emoglobina entro 1 mese. Gli effetti collaterali (che si hanno in circa un quarto dei pazienti) sono costipazione/diarrea, crampi addominali e nausea. Per via parenterale si procede per pazienti con una grave anemia, con malassorbimento o intolleranza per bocca; si possono utilizzare due formulazioni differenti. Il ferro-destrano: [Ferrinject] permette in una sola solamministrazione di ripristinare i livelli di ferro nel corpo; la quota da somministrare è pari a
emoglobina mancante [mg/dl] * peso corporeo [Kg] * 1000-600 mg [M-F]
Gli effetti collaterali sono l’anafilassi (se somministrato rapidamente), le artralgie/mialgie, la febbre, il prurito e/o le linfoadenopatie. Tali reazioni si sono dimostrate di intensità e frequenza tali che attualmente il farmaco è sotto analisi da parte della FDA; sicuramente non sembra legato ad un’allergia al ferro quando alla velocità di somministrazione del ferro stesso. Un’altra terapia é il sodio ferrico gluconato [Venofer] IV che si utilizza soprattutto per i pazienti intolleranti al Ferro-Destrano, non si somministra monodose (per il fore rischio di ipotensione arteriosa) e si somministra a 125 mg in 100 ml di NaCl 0.9% IV in un’ora; viene ripetuta fino al raggiungimento della dose necessaria.
(continua…)
REFERENCES:
1. Liano F, Garcia-Martin F, Gallego A, et al.: Easy and early prognosis in acute tubular necrosis: A forward analysis of 228 cases. Nephron 1989;51:307-313
2. Vincent JL, Baron J-F, Reinhart K, et al.: Anemia and blood transfusion in critically ill patients. JAMA 2002;288:1499-1507
3. Wilkerson DK, Rosen AL, Sehgal LR, et al.: Limits of cardiac compensation in anemic baboons. Surgery 1988;103:665-670
4. Leung JM, Weiskopf RB, Feiner J, et al.: Electrocardiographic ST-segment changes during acute, severe isovolemic hemodilution in humans. Anesthesiology 2000;93:1004-1010
5. Expert Working Group: Guidelines for red blood cell and plasma transfusion for adults and children: Report of the expert working group. Can Med Assoc J 1997;156 (Suppl 11):S1-S24
6. Bayer WL, Coenen WM, Jenkins DC, Zucker ML: The use of blood and blood components in 1,769 patients undergoing open-heart surgery. Ann Thorac Surg 1980;29:117-122
7. Viele MK, Weiskopf RB: What can we learn about the need for transfusion from patients who refuse blood? The experience with Jehovah’s Witnesses. Transfusion 1994;34:396-401
8. Graves CL, Allen RM: Anesthesia in the presence of severe anemia. Rocky Mountain Med J. 1974;67:35-40
9. Slawson KB: Anaesthesia for the patient in renal failure. Br J Anaesth 1972;44:277-282
10. Aldrete JA, Daniel W, O’Higghins JW, et al.: Analysis of anesthetic-related morbidity in human recipients of renal homografts. Anesth Analg 1971;50:321-329
11. Samuel JR, Powell D: Renal transplantation: Anesthetic experience of 100 cases. Anesthesia 1970;125:165-176
12. Gollub S, Bailey CP: Management of major surgical blood loss without transfusion. JAMA 1966;198:149-152
13. Carson JL, Spence RK, Poses RM, Bonavita G: Severity of anaemia and operative mortality and morbidity. Lancet 1988;1:727-729
14. Spence RK, Carson JA, Poses R, et al.: Elective surgery without transfusion: Influence of preoperative hemoglobin level and blood loss on mortality. Am J Surg 1990;159:320-324
15. Ott DA, Cooley DA: Cardiovascular surgery in Jehovah’s Witnesses: Report of 542 operations without blood transfusion. JAMA 1977;238:1256-1258
16. Fullerton WT, Turner AG: Exchange transfusion in treatment of severe anaemia in pregnancy. Lancet 1962;282:75-78
17. Alexiu O, Mircea N, Balaban M, Furtunescu B: Gastro-intestinal haemorrhage from peptic ulcer: An evaluation of bloodless transfusion and early surgery. Anesthesia 975;130:609-615
18. Audet AM, Goodnough LT: Practice strategies for elective red blood cell transfusion. Ann Intern Med 1992;116:403-406
19. American Society of Anesthesiologists Task Force on Blood Component Therapy: Practice guidelines for blood component therapy. Anesthesiology 1996;84:732-747
20. Consensus Conference (National Institutes of Health): Perioperative red blood cell transfusion. JAMA 1988;260:2700-2703
21. Welch HG, Meehan KR, Goodnough LT: Prudent strategies for elective red blood cell transfusion. Ann Intern Med 1992;116:393-402
22. Cane RD: Hemoglobin: How much is enough? Crit Care Med 1990;18:1046-1047
REFERENCES:
1. Liano F, Garcia-Martin F, Gallego A, et al.: Easy and early prognosis in acute tubular necrosis: A forward analysis of 228 cases. Nephron 1989;51:307-313
2. Vincent JL, Baron J-F, Reinhart K, et al.: Anemia and blood transfusion in critically ill patients. JAMA 2002;288:1499-1507
3. Wilkerson DK, Rosen AL, Sehgal LR, et al.: Limits of cardiac compensation in anemic baboons. Surgery 1988;103:665-670
4. Leung JM, Weiskopf RB, Feiner J, et al.: Electrocardiographic ST-segment changes during acute, severe isovolemic hemodilution in humans. Anesthesiology 2000;93:1004-1010
5. Expert Working Group: Guidelines for red blood cell and plasma transfusion for adults and children: Report of the expert working group. Can Med Assoc J 1997;156 (Suppl 11):S1-S24
6. Bayer WL, Coenen WM, Jenkins DC, Zucker ML: The use of blood and blood components in 1,769 patients undergoing open-heart surgery. Ann Thorac Surg 1980;29:117-122
7. Viele MK, Weiskopf RB: What can we learn about the need for transfusion from patients who refuse blood? The experience with Jehovah’s Witnesses. Transfusion 1994;34:396-401
8. Graves CL, Allen RM: Anesthesia in the presence of severe anemia. Rocky Mountain Med J. 1974;67:35-40
9. Slawson KB: Anaesthesia for the patient in renal failure. Br J Anaesth 1972;44:277-282
10. Aldrete JA, Daniel W, O’Higghins JW, et al.: Analysis of anesthetic-related morbidity in human recipients of renal homografts. Anesth Analg 1971;50:321-329
11. Samuel JR, Powell D: Renal transplantation: Anesthetic experience of 100 cases. Anesthesia 1970;125:165-176
12. Gollub S, Bailey CP: Management of major surgical blood loss without transfusion. JAMA 1966;198:149-152
13. Carson JL, Spence RK, Poses RM, Bonavita G: Severity of anaemia and operative mortality and morbidity. Lancet 1988;1:727-729
14. Spence RK, Carson JA, Poses R, et al.: Elective surgery without transfusion: Influence of preoperative hemoglobin level and blood loss on mortality. Am J Surg 1990;159:320-324
15. Ott DA, Cooley DA: Cardiovascular surgery in Jehovah’s Witnesses: Report of 542 operations without blood transfusion. JAMA 1977;238:1256-1258
16. Fullerton WT, Turner AG: Exchange transfusion in treatment of severe anaemia in pregnancy. Lancet 1962;282:75-78
17. Alexiu O, Mircea N, Balaban M, Furtunescu B: Gastro-intestinal haemorrhage from peptic ulcer: An evaluation of bloodless transfusion and early surgery. Anesthesia 975;130:609-615
18. Audet AM, Goodnough LT: Practice strategies for elective red blood cell transfusion. Ann Intern Med 1992;116:403-406
19. American Society of Anesthesiologists Task Force on Blood Component Therapy: Practice guidelines for blood component therapy. Anesthesiology 1996;84:732-747
20. Consensus Conference (National Institutes of Health): Perioperative red blood cell transfusion. JAMA 1988;260:2700-2703
21. Welch HG, Meehan KR, Goodnough LT: Prudent strategies for elective red blood cell transfusion. Ann Intern Med 1992;116:393-402
22. Cane RD: Hemoglobin: How much is enough? Crit Care Med 1990;18:1046-1047
23. Crosby ET: Perioperative haemotherapy: I. Indications for blood component transfusion. Can J Anesth 1992;39:695-707
24. Nelson AH, Fleisher LA, Rosenbaum SH: Relationship between postoperative anemia and cardiac morbidity in high-risk vascular patients in the intensive care unit. Crit Care Med 1993;21:860-866
25. Carson JL, Duff A, Poses RM, et al.: Effects of anaemia and cardiovascular disease on surgical mortality and morbidity. Lancet 1996;48:1055-1060
26. Hebert PC, Wells G, Tweeddale M, et al.: Does transfusion practice affect mortality in critically ill patients? Am J Respir Crit Care Med 1997;155:1618-1623


0 Response to "Anemia in ICU - introduzione generale (Capitolo 2.10b.1-1)"
Posting Komentar